Division of Molecular Medicine, Leeds Institute of Medical Research, University of Leeds, Leeds, West Yorkshire, UK.
Yorkshire Clinical Genetics Service, Chapel Allerton Hospital, Leeds, West Yorkshire, UK.
J Med Genet. 2021 May;58(5):334-341. doi: 10.1136/jmedgenet-2020-106873. Epub 2020 Jun 22.
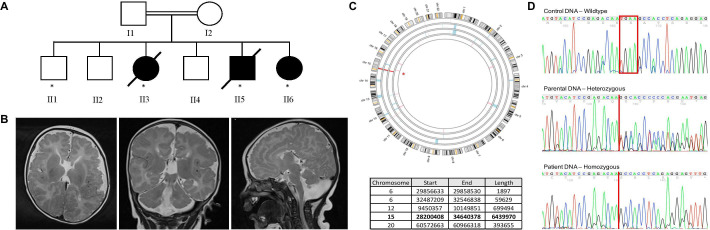
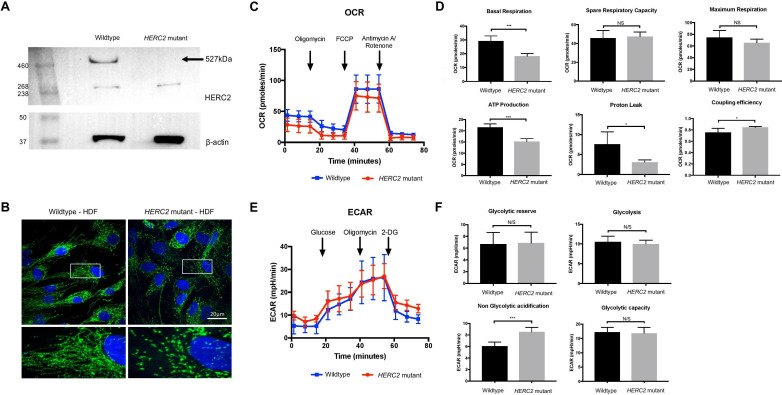
The gene encodes a 527 kDa E3 ubiquitin protein ligase that has key roles in cell cycle regulation, spindle formation during mitosis, mitochondrial functions and DNA damage responses. It has essential roles during embryonic development, particularly for neuronal and muscular functions. To date, missense mutations in have been associated with an autosomal recessive neurodevelopmental disorder with some phenotypical similarities to Angelman syndrome, and a homozygous deletion spanning and causing a more severe neurodevelopmental phenotype.
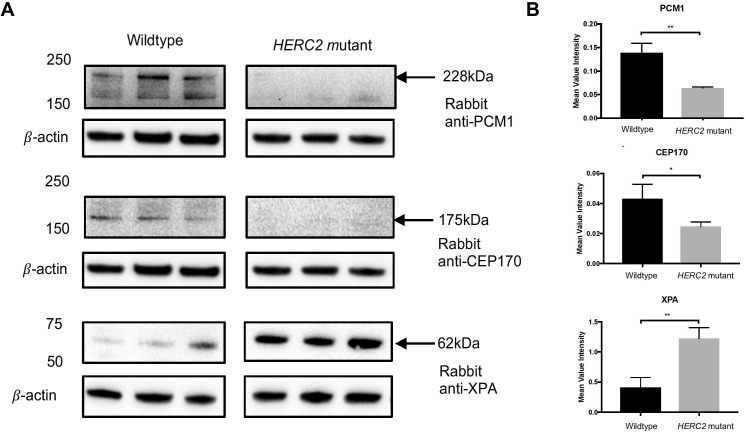
We ascertained a consanguineous family with a presumed autosomal recessive severe neurodevelopmental disorder that leads to paediatric lethality. In affected individuals, we identified a homozygous frameshift variant that results in a premature stop codon and complete loss of HERC2 protein. Functional characterisation of this variant in fibroblasts, from one living affected individual, revealed impaired mitochondrial network and function as well as disrupted levels of known interacting proteins such as XPA.
This study extends the genotype-phenotype correlation for variants to include a distinct lethal neurodevelopmental disorder, highlighting the importance of further characterisation for -related disorders.
该基因编码一种 527kDa 的 E3 泛素蛋白连接酶,在细胞周期调控、有丝分裂纺锤体形成、线粒体功能和 DNA 损伤反应中具有关键作用。它在胚胎发育中起着至关重要的作用,特别是对神经元和肌肉功能。迄今为止,已经发现 中的错义突变与常染色体隐性神经发育障碍有关,其表型与 Angelman 综合征有一些相似之处,而横跨 和 的纯合缺失导致更严重的神经发育表型。
我们确定了一个具有假定常染色体隐性严重神经发育障碍的近亲家族,该障碍导致儿科致死率。在受影响的个体中,我们发现了一种纯合的 HERC2 移码变异,导致提前出现终止密码子和 HERC2 蛋白完全缺失。对来自一名存活受影响个体的成纤维细胞中的这种变体进行功能表征,发现线粒体网络和功能受损,以及已知相互作用蛋白(如 XPA)的水平失调。
本研究将 变体的基因型-表型相关性扩展到包括一种独特的致死性神经发育障碍,突出了进一步对与 HERC2 相关的疾病进行特征描述的重要性。