Hollstein Ronja, Parry David A, Nalbach Lisa, Logan Clare V, Strom Tim M, Hartill Verity L, Carr Ian M, Korenke Georg C, Uppal Sandeep, Ahmed Mushtaq, Wieland Thomas, Markham Alexander F, Bennett Christopher P, Gillessen-Kaesbach Gabriele, Sheridan Eamonn G, Kaiser Frank J, Bonthron David T
Sektion für Funktionelle Genetik am Institut für Humangenetik, Universität zu Lübeck, Lübeck, Germany.
Section of Genetics, School of Medicine, University of Leeds, Leeds, UK.
J Med Genet. 2015 Dec;52(12):797-803. doi: 10.1136/jmedgenet-2015-103344. Epub 2015 Sep 30.
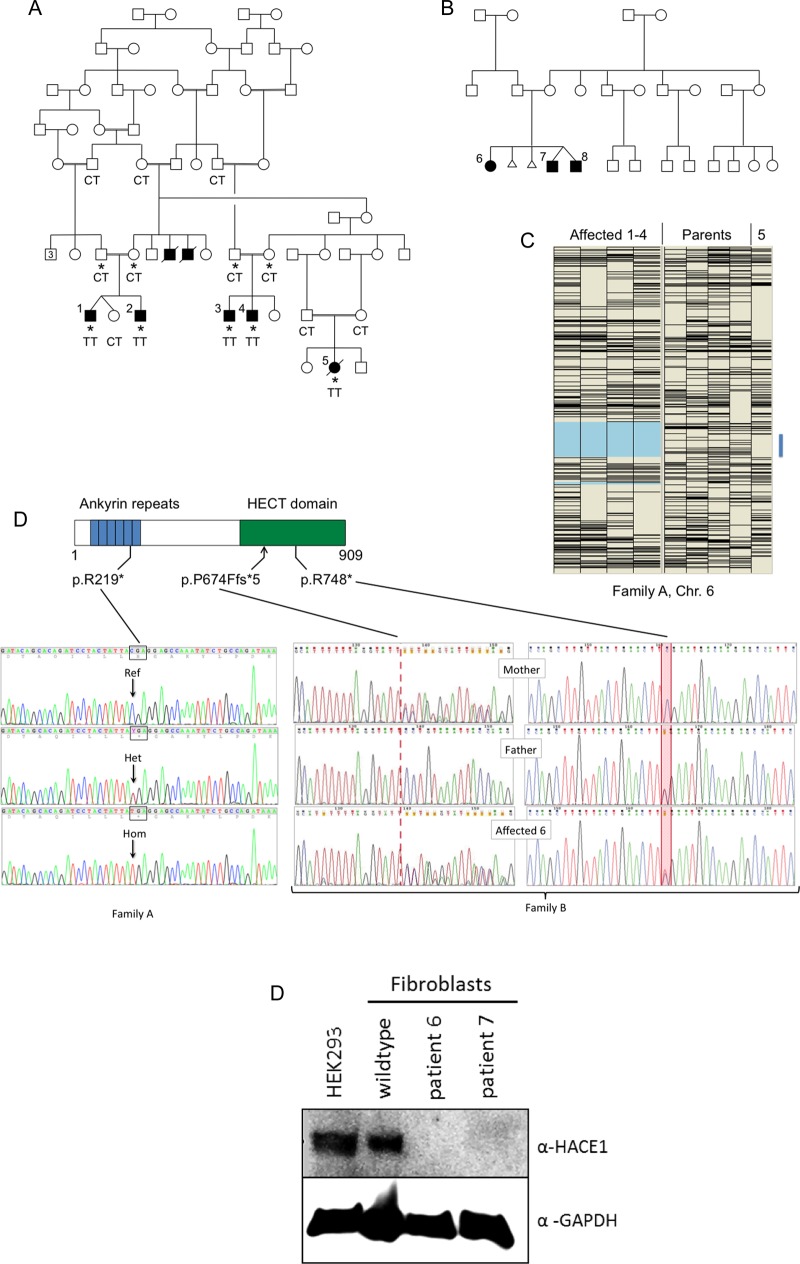
The genetic aetiology of neurodevelopmental defects is extremely diverse, and the lack of distinctive phenotypic features means that genetic criteria are often required for accurate diagnostic classification. We aimed to identify the causative genetic lesions in two families in which eight affected individuals displayed variable learning disability, spasticity and abnormal gait.
Autosomal recessive inheritance was suggested by consanguinity in one family and by sibling recurrences with normal parents in the second. Autozygosity mapping and exome sequencing, respectively, were used to identify the causative gene.
In both families, biallelic loss-of-function mutations in HACE1 were identified. HACE1 is an E3 ubiquitin ligase that regulates the activity of cellular GTPases, including Rac1 and members of the Rab family. In the consanguineous family, a homozygous mutation p.R219* predicted a truncated protein entirely lacking its catalytic domain. In the other family, compound heterozygosity for nonsense mutation p.R748* and a 20-nt insertion interrupting the catalytic homologous to the E6-AP carboxyl terminus (HECT) domain was present; western blot analysis of patient cells revealed an absence of detectable HACE1 protein.
HACE1 mutations underlie a new autosomal recessive neurodevelopmental disorder. Previous studies have implicated HACE1 as a tumour suppressor gene; however, since cancer predisposition was not observed either in homozygous or heterozygous mutation carriers, this concept may require re-evaluation.
神经发育缺陷的遗传病因极为多样,且缺乏独特的表型特征意味着准确的诊断分类通常需要遗传标准。我们旨在确定两个家族中的致病基因损伤,这两个家族中有8名受影响个体表现出不同程度的学习障碍、痉挛和异常步态。
一个家族中通过近亲结婚提示常染色体隐性遗传,另一个家族中通过正常父母的同胞复发提示常染色体隐性遗传。分别使用纯合子定位和外显子组测序来确定致病基因。
在两个家族中均鉴定出HACE1基因的双等位基因功能丧失突变。HACE1是一种E3泛素连接酶,可调节细胞GTP酶的活性,包括Rac1和Rab家族成员。在近亲家族中,纯合突变p.R219预测会产生一种完全缺乏催化结构域的截短蛋白。在另一个家族中,存在无义突变p.R748的复合杂合性以及一个20个核苷酸的插入,该插入中断了与E6-AP羧基末端(HECT)结构域同源的催化结构域;对患者细胞的蛋白质印迹分析显示未检测到HACE1蛋白。
HACE1突变是一种新的常染色体隐性神经发育障碍的基础。先前的研究表明HACE1是一种肿瘤抑制基因;然而,由于在纯合或杂合突变携带者中均未观察到癌症易感性,这一概念可能需要重新评估。