Laboratory of Complex Biological Systems and Bioinformatics (CBB), Department of Bioinformatics, Institute of Biochemistry and Biophysics (IBB), University of Tehran, Tehran, Iran.
Department of Immunology, School of Medicine, Tehran University of Medical Sciences, Tehran, Iran.
Arthritis Res Ther. 2020 Jun 23;22(1):156. doi: 10.1186/s13075-020-02239-3.
A comprehensive intuition of the systemic lupus erythematosus (SLE), as a complex and multifactorial disease, is a biological challenge. Dealing with this challenge needs employing sophisticated bioinformatics algorithms to discover the unknown aspects. This study aimed to underscore key molecular characteristics of SLE pathogenesis, which may serve as effective targets for therapeutic intervention.
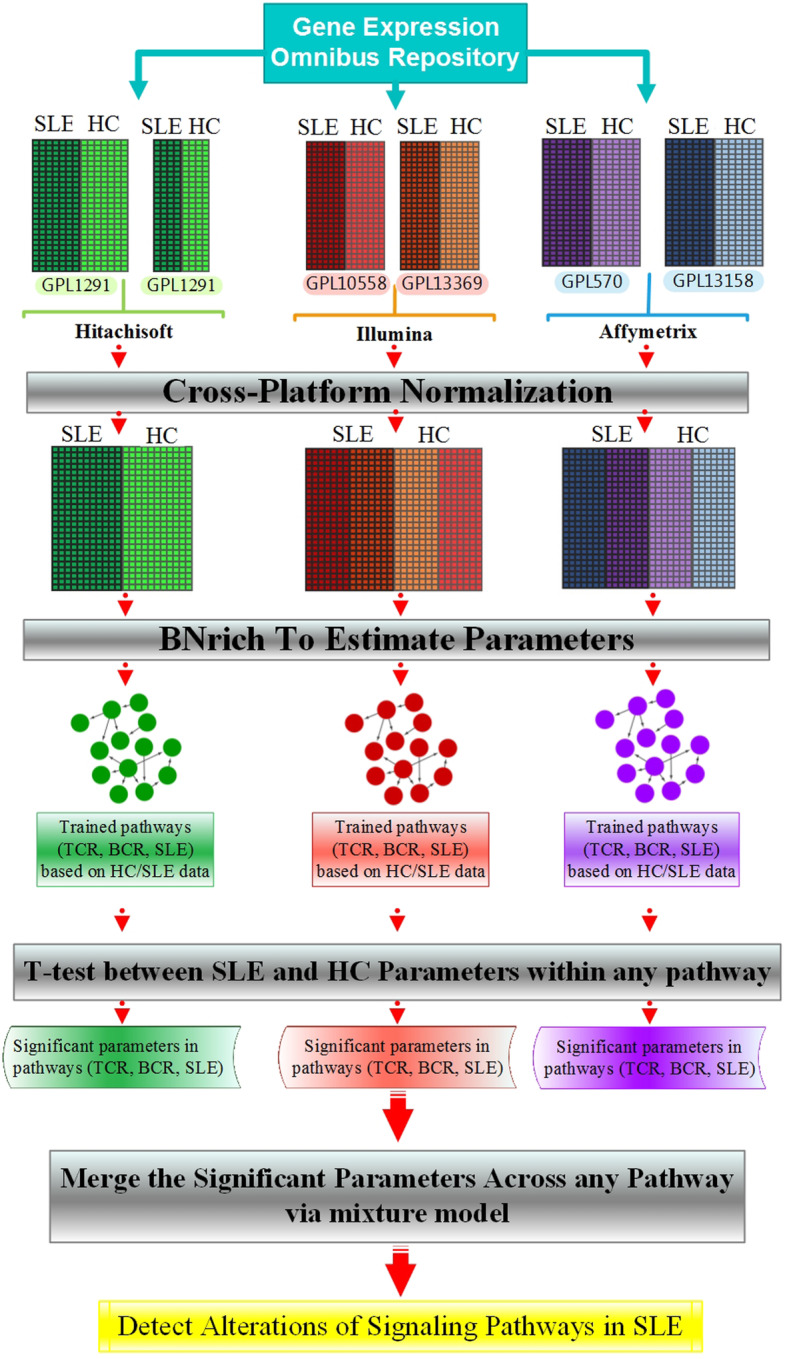
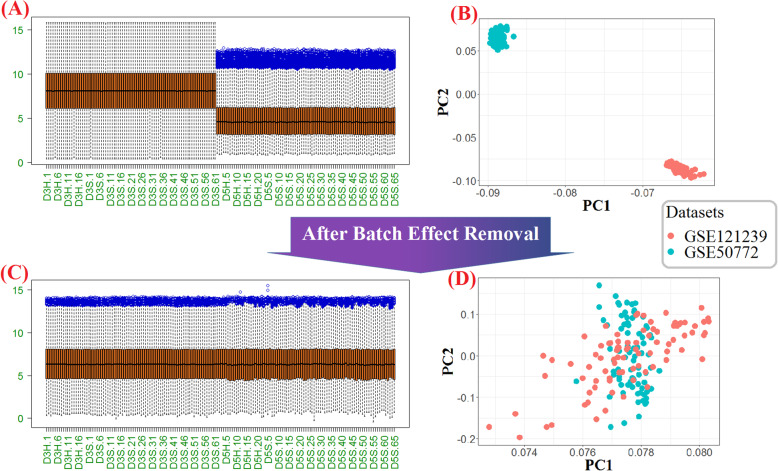
In the present study, the human peripheral blood mononuclear cell (PBMC) microarray datasets (n = 6), generated by three platforms, which included SLE patients (n = 220) and healthy control samples (n = 135) were collected. Across each platform, we integrated the datasets by cross-platform normalization (CPN). Subsequently, through BNrich method, the structures of Bayesian networks (BNs) were extracted from KEGG-indexed SLE, TCR, and BCR signaling pathways; the values of the node (gene) and edge (intergenic relationships) parameters were estimated within each integrated datasets. Parameters with the FDR < 0.05 were considered significant. Finally, a mixture model was performed to decipher the signaling pathway alterations in the SLE patients compared to healthy controls.
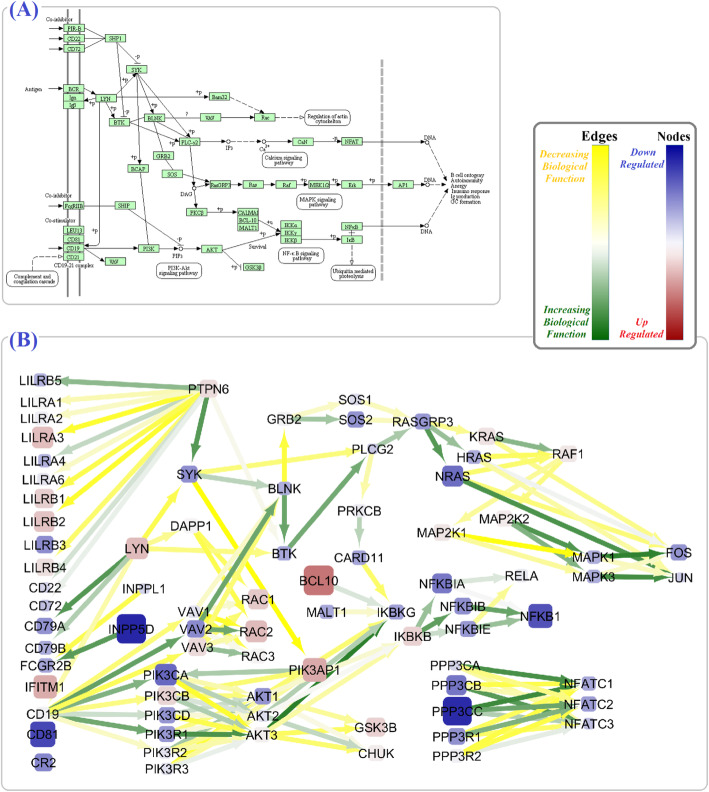
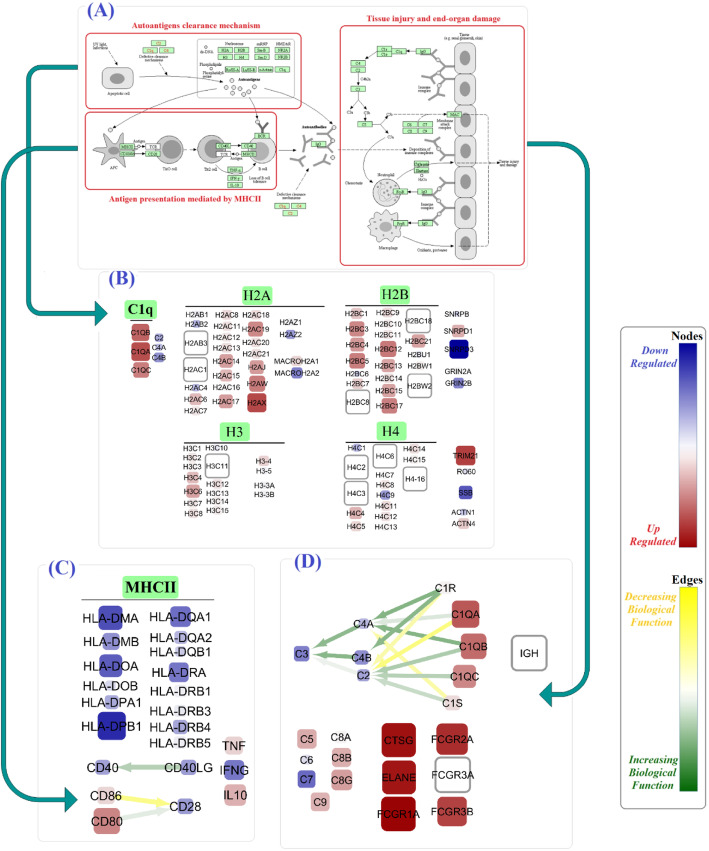
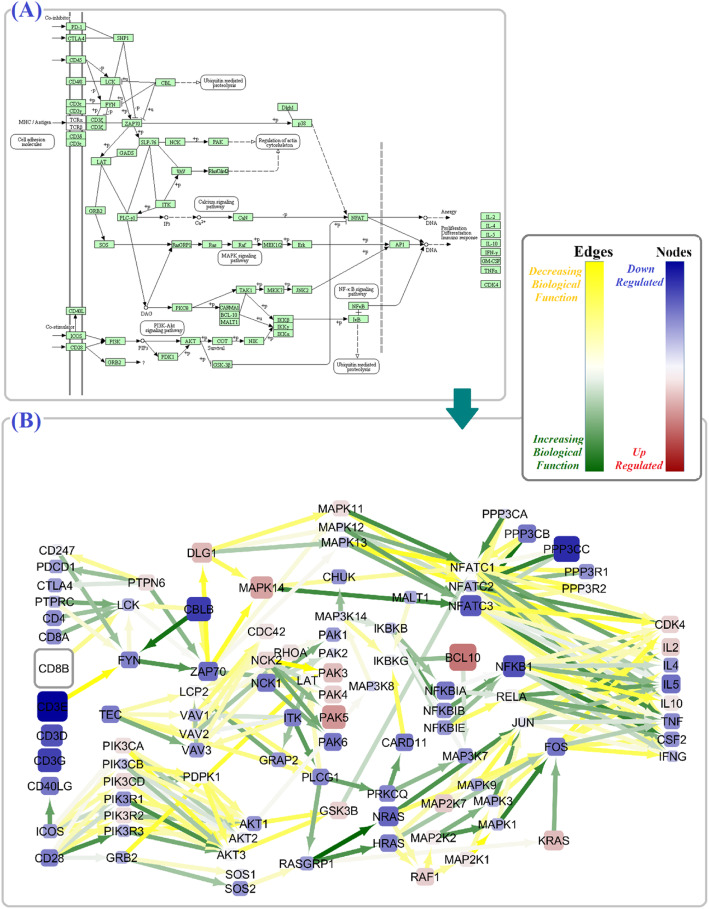
In the SLE signaling pathway, we identified the dysregulation of several nodes involved in the (1) clearance mechanism (SSB, MACROH2A2, TRIM21, H2AX, and C1Q gene family), (2) autoantigen presentation by MHCII (HLA gene family, CD80, IL10, TNF, and CD86), and (3) end-organ damage (FCGR1A, ELANE, and FCGR2A). As a remarkable finding, we demonstrated significant perturbation in CD80 and CD86 to CD28, CD40LG to CD40, C1QA and C1R to C2, and C1S to C4A edges. Moreover, we not only replicated previous studies regarding alterations of subnetworks involved in TCR and BCR signaling pathways (PI3K/AKT, MAPK, VAV gene family, AP-1 transcription factor) but also distinguished several significant edges between genes (PPP3 to NFATC gene families). Our findings unprecedentedly showed that different parameter values assign to the same node based on the pathway topology (the PIK3CB parameter values were 1.7 in TCR vs - 0.5 in BCR signaling pathway).
Applying the BNrich as a hybridized network construction method, we highlight under-appreciated systemic alterations of SLE, TCR, and BCR signaling pathways in SLE. Consequently, having such a systems biology approach opens new insights into the context of multifactorial disorders.
全面了解系统性红斑狼疮(SLE)作为一种复杂的多因素疾病是一个生物学挑战。应对这一挑战需要采用复杂的生物信息学算法来发现未知的方面。本研究旨在强调 SLE 发病机制的关键分子特征,这些特征可能成为治疗干预的有效靶点。
本研究收集了三个平台生成的人类外周血单个核细胞(PBMC)微阵列数据集(n=6),其中包括 SLE 患者(n=220)和健康对照样本(n=135)。在每个平台上,我们通过跨平台归一化(CPN)整合数据集。随后,通过 BNrich 方法,从 KEGG 索引的 SLE、TCR 和 BCR 信号通路中提取贝叶斯网络(BN)的结构;在每个集成数据集中估计节点(基因)和边缘(基因间关系)参数的值。FDR<0.05 的参数被认为是显著的。最后,采用混合模型来破译 SLE 患者与健康对照组之间信号通路的改变。
在 SLE 信号通路中,我们发现几个参与(1)清除机制(SSB、MACROH2A2、TRIM21、H2AX 和 C1Q 基因家族)、(2)MHCII 介导的自身抗原呈递(HLA 基因家族、CD80、IL10、TNF 和 CD86)和(3)终末器官损伤(FCGR1A、ELANE 和 FCGR2A)的节点失调。值得注意的是,我们证明了 CD80 和 CD86 与 CD28、CD40LG 与 CD40、C1QA 和 C1R 与 C2、C1S 与 C4A 边缘的显著干扰。此外,我们不仅复制了先前关于 TCR 和 BCR 信号通路(PI3K/AKT、MAPK、VAV 基因家族、AP-1 转录因子)中涉及的亚网络改变的研究,而且还区分了基因之间的几个显著边缘(PPP3 到 NFATC 基因家族)。我们的发现前所未有地表明,基于通路拓扑,相同节点的参数值不同(TCR 中的 PIK3CB 参数值为 1.7,而 BCR 信号通路中的参数值为-0.5)。
应用 BNrich 作为一种混合网络构建方法,我们强调了 SLE、TCR 和 BCR 信号通路中 SLE 被低估的系统性改变。因此,采用这种系统生物学方法为多因素疾病的背景提供了新的见解。