Li Juming, Liu Lingxiang, Zhang Qi, Huang Yumin, Zhang Yihong, Gan Xiaoyan, Liu Siqin, Yue Zhen, Wei Yongzhong
Department of Orthopedics.
Department of Medical Oncology, the First Affiliated Hospital of Nanjing Medical University, Nanjing.
Medicine (Baltimore). 2020 Jun 26;99(26):e20725. doi: 10.1097/MD.0000000000020725.
Malignant peripheral nerve sheath tumor (MPNST) is a rare sarcoma. Owing to the lack of specific histological criteria, immunohistochemical, and molecular diagnostic markers, several differential diagnoses must be considered. Advances in molecular testing can provide significant insights for management of rare tumor.
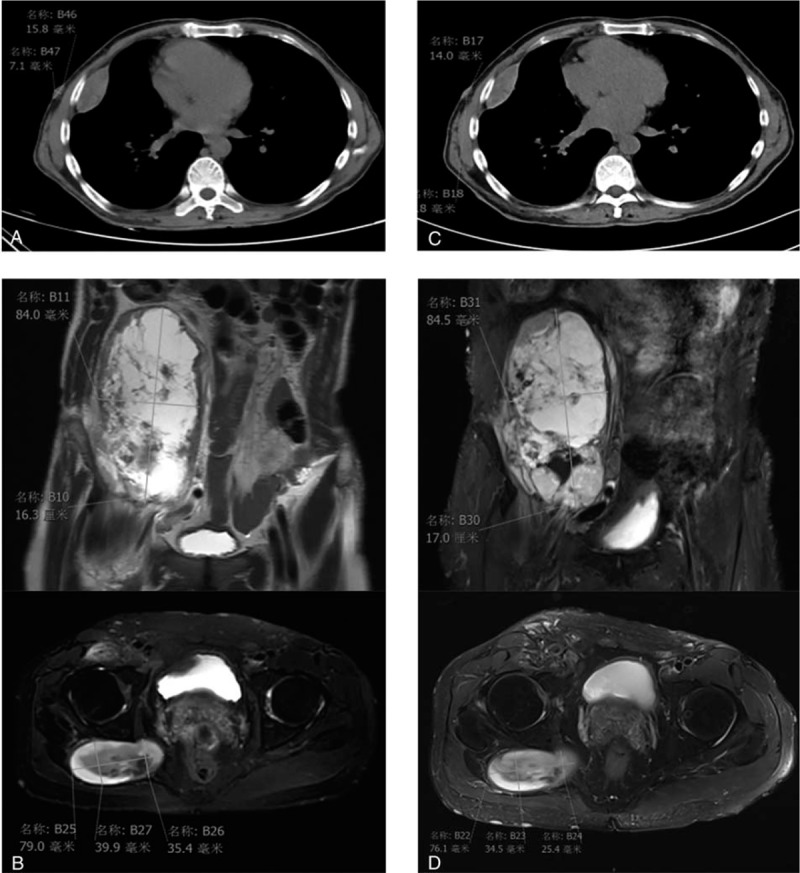
The patient was a 50-year-old man with a history of lumpectomy on the right back 30 years ago. He felt a stabbing pain at the right iliac fossa and went to the local hospital.
By immunohistochemistry, the tumor cells stained positively for S-100 (focal +), CD34 (strong +++) and Ki-67 (20%), and negatively for smooth muscle actin, pan-cytokeratin, neurofilament, pan-cytokeratin-L, GFAP, CD31, STAT6, ERG, myogenin, and MyoD1. Combined with the histopathology and immunohistochemistry results, our initial diagnosis was solitary fibrous tumor (SFT) or MPNST. The tissue biopsy was sent for next-generation sequencing. neurofibromatosis type 1 Q1395Hfs22 somatic mutation, neurofibromatosis type 1 D483Tfs15 germline mutation, and amplifications of BTK, MDM2, ATF1, BMPR1A, EBHA2, GNA13, PTPN11, RAD52, RPTOR, and SOX9, as well as TJP1-ROS1 fusion, CDKN2A-IL1RAPL2 fusion and CDKN2A/UBAP1 rearrangement were identified. Given that NAB2-STAT6 fusion, a specific biomarker of SFT, was not identified in our patient's tumor, the SFT was excluded by through genetic testing results. Therefore, our finally diagnosis was a MPNST by 2 or more pathologists.
Subsequently, the patient received crizotinib therapy for 2 months and showed stable disease. However, after crizotinib continued treatment for 4 months, the patient's disease progressed. Soon after, the patient stopped crizotinib treatment and died in home.
To our knowledge, this is the first report of the TJP1-ROS1 fusion, which expands the list of gene fusions and highlights new targets for targeted therapy. Also, our case underlines the value of multi-gene panel next-generation sequencing for diagnosis of MPNST.
恶性外周神经鞘瘤(MPNST)是一种罕见的肉瘤。由于缺乏特异性组织学标准、免疫组化及分子诊断标志物,必须考虑多种鉴别诊断。分子检测的进展可为罕见肿瘤的管理提供重要见解。
该患者为一名50岁男性,30年前有右背部肿块切除术史。他感到右髂窝刺痛,遂前往当地医院就诊。
通过免疫组化,肿瘤细胞S-100呈阳性(局灶性+)、CD34呈强阳性(+++)、Ki-67呈阳性(20%),而平滑肌肌动蛋白、全细胞角蛋白、神经丝、全细胞角蛋白-L、胶质纤维酸性蛋白、CD31、信号转导和转录激活因子6(STAT6)、红细胞生成素受体(ERG)、生肌调节因子、肌分化抗原1呈阴性。结合组织病理学和免疫组化结果,我们初步诊断为孤立性纤维瘤(SFT)或MPNST。将组织活检样本送去进行二代测序。检测到1型神经纤维瘤病Q1395Hfs22体细胞突变、1型神经纤维瘤病D483Tfs15种系突变,以及布鲁顿酪氨酸激酶(BTK)、小鼠双微体2(MDM2)、活化转录因子1(ATF1)、骨形态发生蛋白受体1A(BMPR1A)、EBHA2、鸟嘌呤核苷酸结合蛋白α13亚基(GNA13)、蛋白酪氨酸磷酸酶非受体型11(PTPN11)、RAD52修复蛋白(RAD52)、含脯氨酸富亮氨酸重复序列的TOR(RPTOR)和SRY-盒转录因子9(SOX9)的扩增,以及紧密连接蛋白1(TJP1)-ROS1融合、细胞周期蛋白依赖性激酶抑制剂2A(CDKN2A)-白细胞介素1受体相关蛋白2(IL1RAPL2)融合和CDKN2A/泛素相关蛋白1(UBAP1)重排。鉴于未在患者肿瘤中检测到SFT的特异性生物标志物NAB2-STAT6融合,通过基因检测结果排除了SFT。因此,2名或更多病理学家最终诊断为MPNST。
随后患者接受克唑替尼治疗2个月,病情稳定。然而,克唑替尼继续治疗4个月后,患者病情进展。此后不久,患者停止克唑替尼治疗,在家中死亡。
据我们所知,这是关于TJP1-ROS1融合的首次报告,它扩展了基因融合列表,并突出了靶向治疗的新靶点。此外我们的病例强调了多基因二代测序在MPNST诊断中的价值。