Department of Neonatology, Neonatal Biophysical Monitoring and Cardiopulmonary Therapies Research Unit, Poznan University of Medical Sciences, Poznan, Poland.
Department of Pathology, Poznan University of Medical Sciences and Greater Poland Cancer Center, Poznan, Poland.
BMC Pediatr. 2020 Jun 29;20(1):320. doi: 10.1186/s12887-020-02200-y.
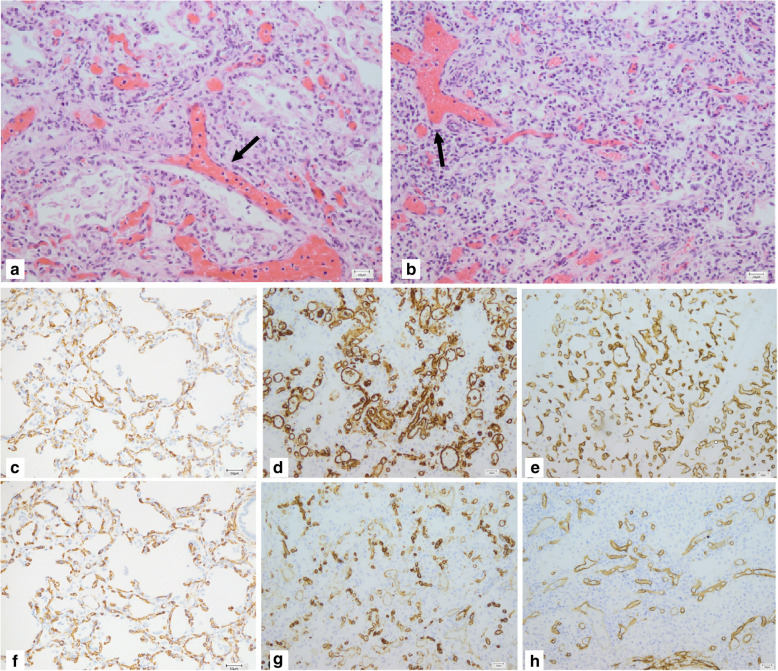
Alveolar capillary dysplasia (ACD) is a rare cause of severe pulmonary hypertension and respiratory failure in neonates. The onset of ACD is usually preceded by a short asymptomatic period. The condition is refractory to all available therapies as it irreversibly affects development of the capillary bed in the lungs. The diagnosis of ACD is based on histopathological evaluation of lung biopsy or autopsy tissue or genetic testing of FOXF1 on chromosome 16q24.1. Here, we describe the first two Polish patients with ACD confirmed by histopathological and genetic examination.
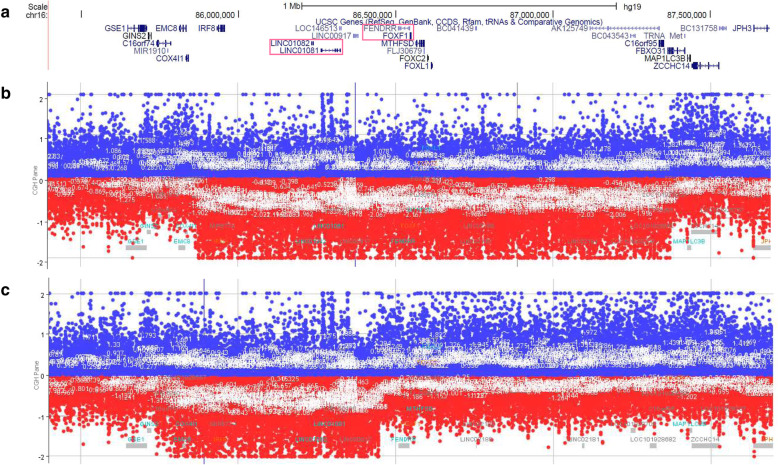
The patients were term neonates with high Apgar scores in the first minutes of life. They both were diagnosed prenatally with heart defects. Additionally, the first patient presented with omphalocele. The neonate slightly deteriorated around 12 hour of life, but underwent surgical repair of omphalocele followed by mechanical ventilation. Due to further deterioration, therapy included inhaled nitric oxide (iNO), inotropes and surfactant administration. The second patient was treated with prostaglandin E1 since birth due to suspicion of aortic coarctation (CoA). After ruling out CoA in the 3 day of life, infusion of prostaglandin E1 was discountinued and immediately patient's condition worsened. Subsequent treatment included re-administration of prostaglandin E1, iNO and mechanical ventilation. Both patients presented with transient improvement after application of iNO, but died despite maximized therapy. They were histopathologically diagnosed post-mortem with ACD. Array comparative genomic hybridization in patient one and patient two revealed copy-number variant (CNV) deletions, respectively, ~ 1.45 Mb in size involving FOXF1 and an ~ 0.7 Mb in size involving FOXF1 enhancer and leaving FOXF1 intact.
Both patients presented with a distinct course of ACD, extra-pulmonary manifestations and response to medications. Surgery and ceasing of prostaglandin E1 infusion should be considered as potential causes of this variability. We further highlight the necessity of thorough genetic testing and histopathological examination and propose immunostaining for CD31 and CD34 to facilitate the diagnostic process for better management of infants with ACD.
肺泡毛细血管发育不良(ACD)是新生儿严重肺动脉高压和呼吸衰竭的罕见原因。ACD 的发病通常在无症状期后很短的时间内。由于它不可逆地影响肺部毛细血管床的发育,因此对所有现有治疗方法均具有抗性。ACD 的诊断基于肺活检或尸检组织的组织病理学评估,或染色体 16q24.1 上的 FOXF1 基因检测。在这里,我们描述了通过组织病理学和基因检测证实的波兰首例 ACD 患者。
这两名患者均为足月新生儿,在生命最初几分钟的 Apgar 评分均较高。他们在产前均被诊断出患有心脏缺陷。此外,第一例患者还伴有脐膨出。新生儿在生命的 12 小时左右略有恶化,但接受了脐膨出的手术修复,随后进行了机械通气。由于病情进一步恶化,治疗包括吸入一氧化氮(iNO)、正性肌力药和表面活性剂的应用。第二名患者因主动脉缩窄(CoA)的怀疑而从出生起就接受前列腺素 E1 治疗。在出生后第 3 天排除 CoA 后,停止了前列腺素 E1 的输注,随后患者病情恶化。随后的治疗包括重新开始使用前列腺素 E1、iNO 和机械通气。两名患者在使用 iNO 后均出现短暂改善,但尽管进行了最大程度的治疗,最终仍死亡。他们死后的组织病理学诊断为 ACD。患者 1 的阵列比较基因组杂交和患者 2 的阵列比较基因组杂交分别显示了大小约为 1.45Mb 的 FOXF1 缺失和大小约为 0.7Mb 的 FOXF1 增强子缺失,而 FOXF1 保持完整。
两名患者均表现出明显的 ACD 病程、肺外表现和对药物的反应。手术和停止前列腺素 E1 输注应被视为这种变异性的潜在原因。我们进一步强调了进行彻底的基因检测和组织病理学检查的必要性,并提出使用 CD31 和 CD34 的免疫染色来促进诊断过程,以更好地管理患有 ACD 的婴儿。