Department of Bioengineering, Swanson School of Engineering, University of Pittsburgh, Pittsburgh, PA 15213;
UPMC Hillman Cancer Center, University of Pittsburgh Medical Center, Pittsburgh, PA 15232.
Proc Natl Acad Sci U S A. 2020 Jul 14;117(28):16500-16508. doi: 10.1073/pnas.2000648117. Epub 2020 Jun 29.
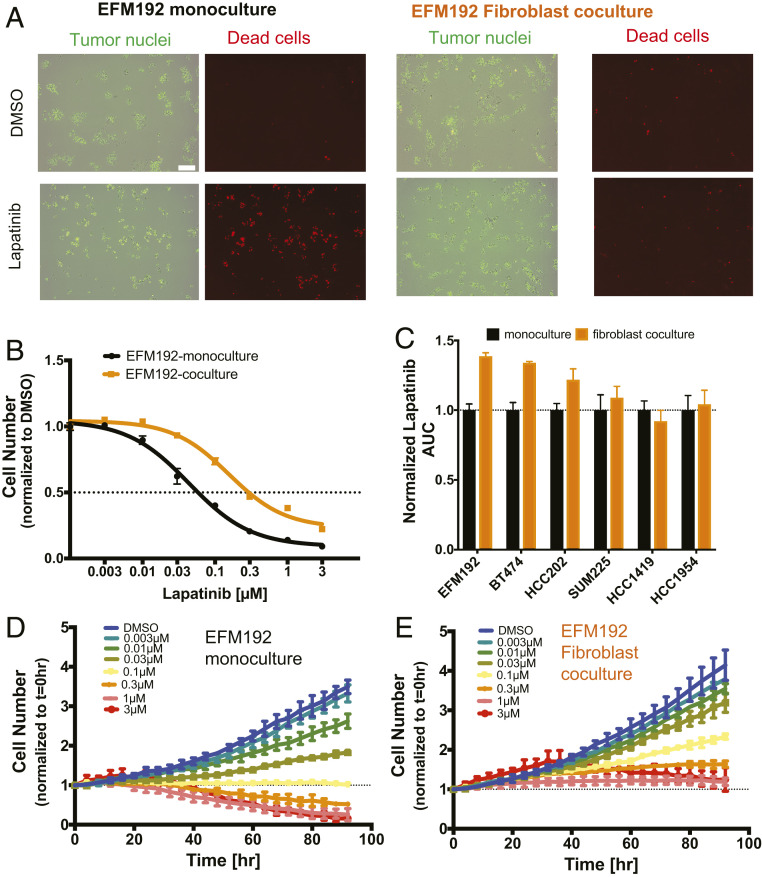
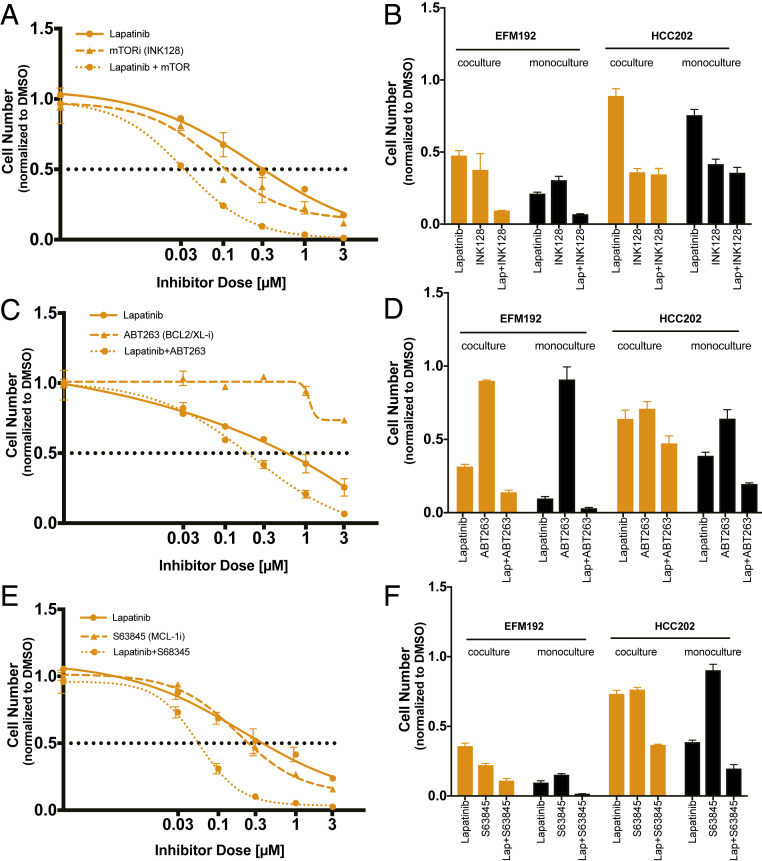
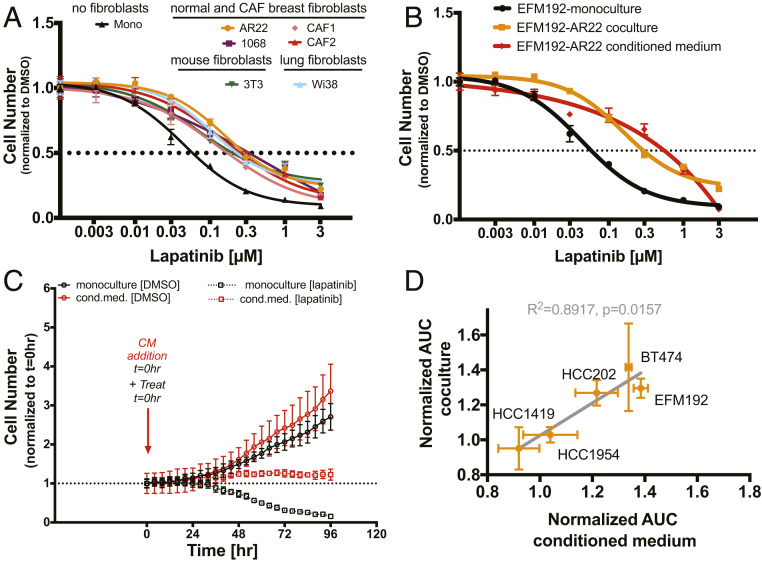
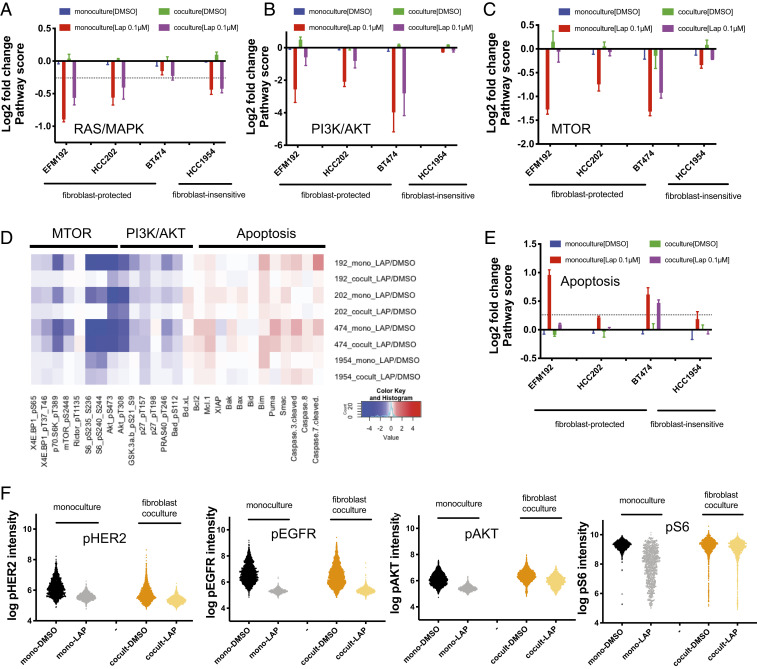
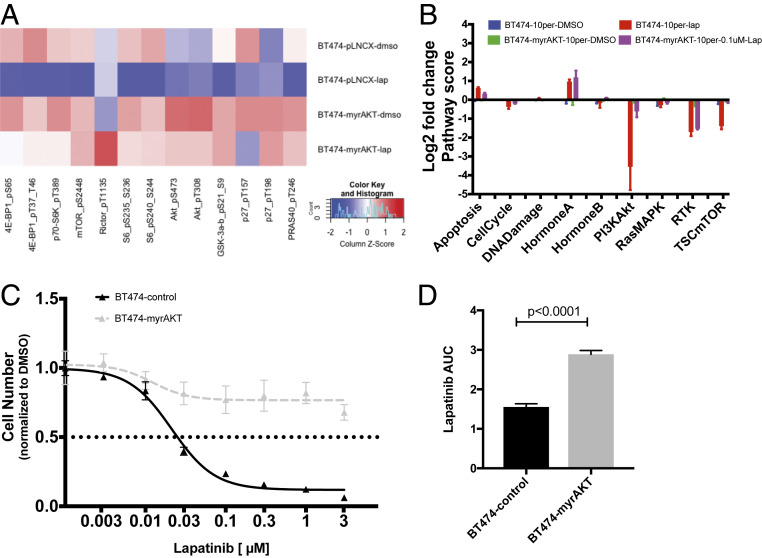
Despite the implementation of multiple HER2-targeted therapies, patients with advanced HER2 breast cancer ultimately develop drug resistance. Stromal fibroblasts represent an abundant cell type in the tumor microenvironment and have been linked to poor outcomes and drug resistance. Here, we show that fibroblasts counteract the cytotoxic effects of HER2 kinase-targeted therapy in a subset of HER2 breast cancer cell lines and allow cancer cells to proliferate in the presence of the HER2 kinase inhibitor lapatinib. Fibroblasts from primary breast tumors, normal breast tissue, and lung tissue have similar protective effects on tumor cells via paracrine factors. This fibroblast-mediated reduction in drug sensitivity involves increased expression of antiapoptotic proteins and sustained activation of the PI3K/AKT/MTOR pathway, despite inhibition of the HER2 and the RAS-ERK pathways in tumor cells. HER2 therapy sensitivity is restored in the fibroblast cocultures by combination treatment with inhibitors of MTOR or the antiapoptotic proteins BCL-XL and MCL-1. Expression of activated AKT in tumor cells recapitulates the effects of fibroblasts resulting in sustained MTOR signaling and poor lapatinib response. Lapatinib sensitivity was not altered by fibroblasts in tumor cells that exhibited sustained MTOR signaling due to a strong gain-of-function PI3KCA mutation. These findings indicate that in addition to tumor cell-intrinsic mechanisms that cause constitutive PI3K/AKT/MTOR pathway activation, secreted factors from fibroblasts can maintain this pathway in the context of HER2 inhibition. Our integrated proteomic-phenotypic approach presents a strategy for the discovery of protective mechanisms in fibroblast-rich tumors and the design of rational combination therapies to restore drug sensitivity.
尽管已经实施了多种针对 HER2 的靶向治疗,但晚期 HER2 乳腺癌患者最终还是会产生耐药性。基质成纤维细胞是肿瘤微环境中丰富的细胞类型之一,与不良结局和耐药性有关。在这里,我们表明成纤维细胞在一组 HER2 乳腺癌细胞系中抵消了 HER2 激酶靶向治疗的细胞毒性作用,并允许癌细胞在 HER2 激酶抑制剂拉帕替尼的存在下增殖。来自原发性乳腺癌、正常乳腺组织和肺组织的成纤维细胞通过旁分泌因子对肿瘤细胞具有相似的保护作用。这种成纤维细胞介导的药物敏感性降低涉及抗凋亡蛋白的表达增加和 PI3K/AKT/MTOR 途径的持续激活,尽管在肿瘤细胞中抑制了 HER2 和 RAS-ERK 途径。在成纤维细胞共培养物中,通过联合使用 MTOR 抑制剂或抗凋亡蛋白 BCL-XL 和 MCL-1 的治疗,恢复了 HER2 治疗的敏感性。肿瘤细胞中激活的 AKT 的表达再现了成纤维细胞的作用,导致持续的 MTOR 信号和拉帕替尼反应不良。在由于强烈的功能获得性 PI3KCA 突变而导致持续 MTOR 信号的肿瘤细胞中,成纤维细胞对拉帕替尼的敏感性没有改变。这些发现表明,除了导致 PI3K/AKT/MTOR 途径持续激活的肿瘤细胞内在机制外,成纤维细胞分泌的因子还可以在 HER2 抑制的情况下维持该途径。我们的综合蛋白质组学-表型方法为发现富含成纤维细胞的肿瘤中的保护机制和设计恢复药物敏感性的合理联合治疗策略提供了一种策略。