Graduate Institute of Clinical Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung 80708, Taiwan.
Division of Rheumatology, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80756, Taiwan.
Int J Mol Sci. 2020 Jul 1;21(13):4702. doi: 10.3390/ijms21134702.
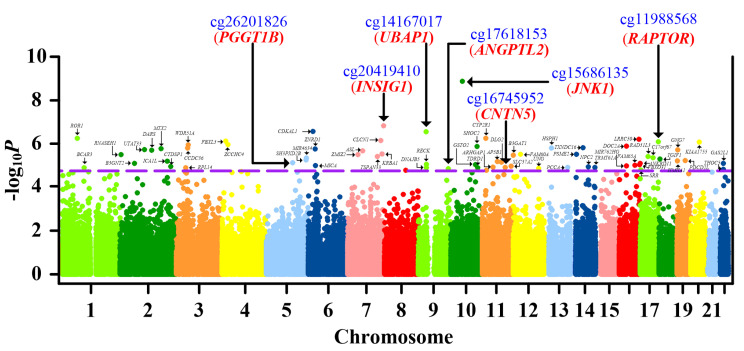
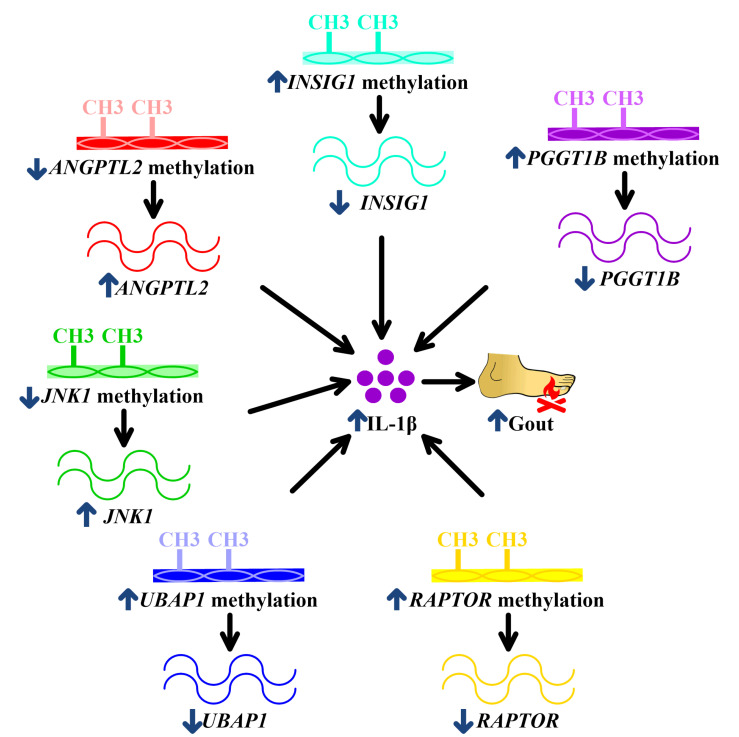
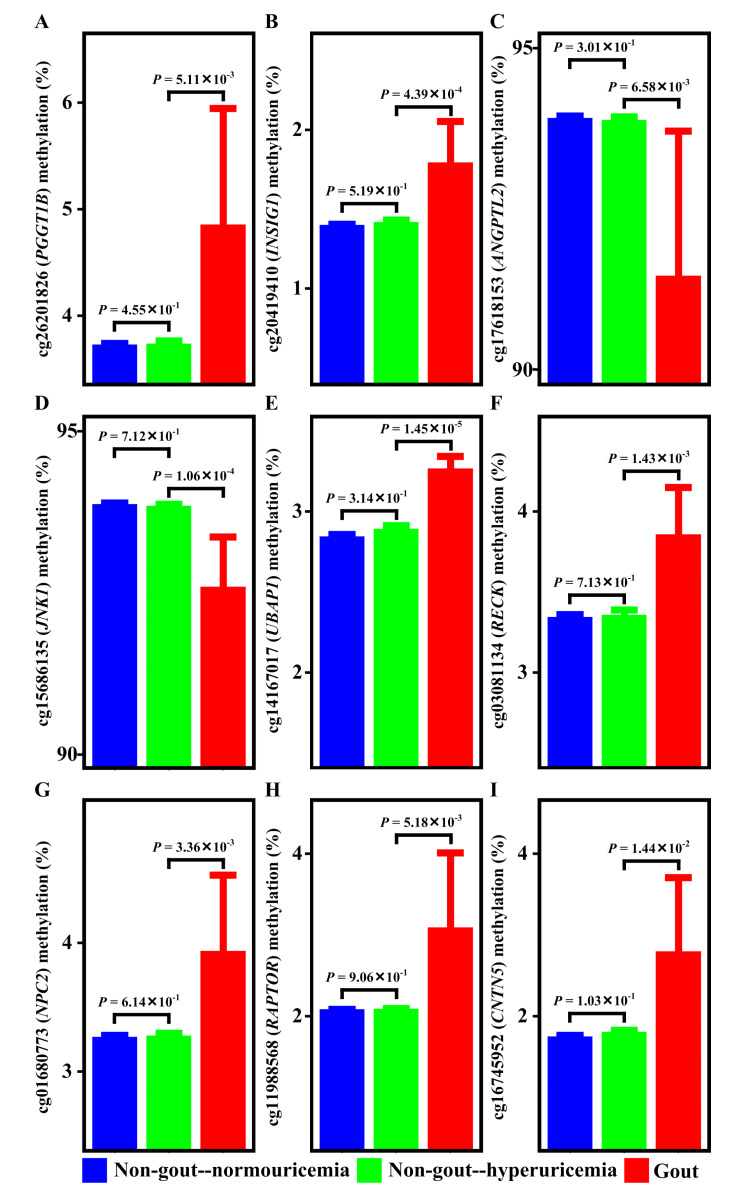
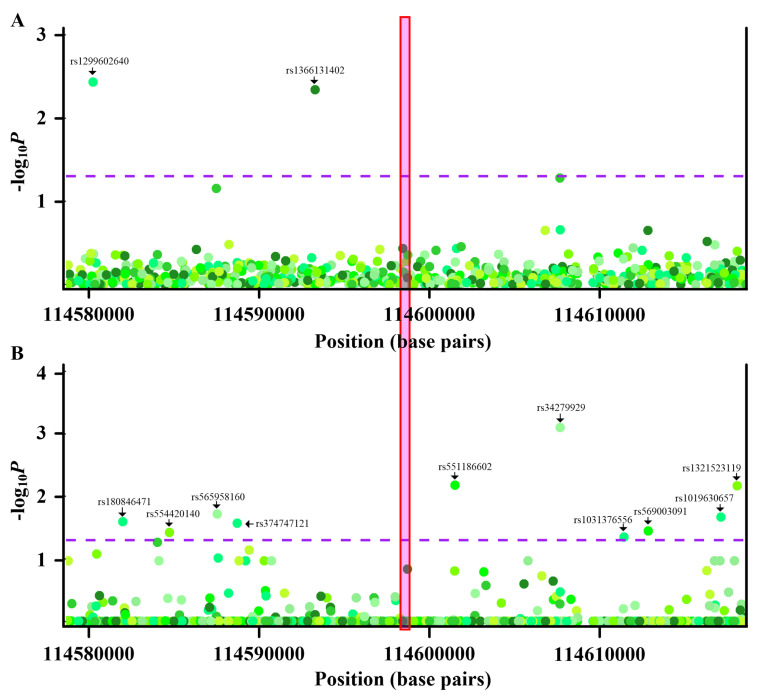
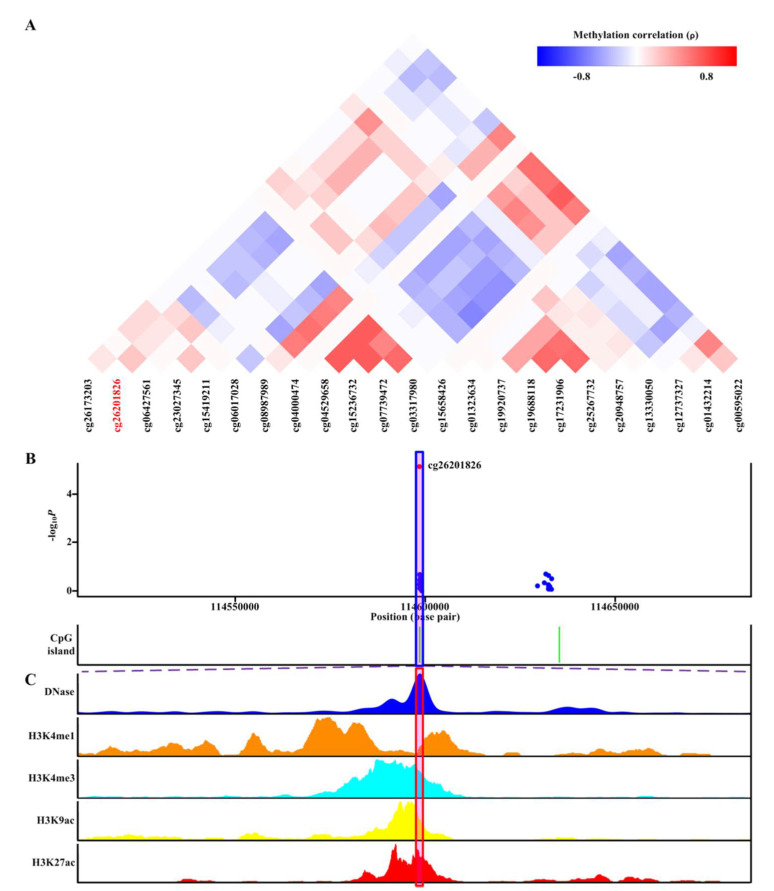
Current knowledge of gout centers on hyperuricemia. Relatively little is known regarding the pathogenesis of gouty inflammation. To investigate the epigenetic background of gouty inflammation independent of hyperuricemia and its relationship to genetics, 69 gout patients and 1455 non-gout controls were included. Promoter-wide methylation was profiled with EPIC array. Whole-genome sequencing data were included for genetic and methylation quantitative trait loci (meQTL) analyses and causal inference tests. Identified loci were subjected to co-methylation analysis and functional localization with DNase hypersensitivity and histone marks analysis. An expression database was queried to clarify biologic functions of identified loci. A transcription factor dataset was integrated to identify transcription factors coordinating respective expression. In total, seven CpG loci involved in interleukin-1β production survived genetic/meQTL analyses, or causal inference tests. None had a significant relationship with various metabolic traits. Additional analysis suggested gouty inflammation, instead of hyperuricemia, provides the link between these CpG sites and gout. Six (, , , , , and ) were novel genes in the field of gout. One () was previously associated with gouty inflammation. Transcription factor mapping identified several potential transcription factors implicated in the link between differential methylation, interleukin-1β production, and gouty inflammation. In conclusion, this study revealed several novel genes specific to gouty inflammation and provided enhanced insight into the biological basis of gouty inflammation.
目前对痛风的认识主要集中在高尿酸血症上。对于痛风炎症的发病机制,人们知之甚少。为了研究与高尿酸血症无关的痛风炎症的表观遗传背景及其与遗传的关系,纳入了 69 名痛风患者和 1455 名非痛风对照者。采用 EPIC 阵列对启动子广泛甲基化进行了分析。全基因组测序数据用于遗传和甲基化数量性状基因座(meQTL)分析和因果关系推断测试。对鉴定出的基因座进行共甲基化分析,并利用 DNase 超敏性和组蛋白标记分析进行功能定位。查询了一个表达数据库,以阐明鉴定出的基因座的生物学功能。整合了一个转录因子数据集,以确定协调各自表达的转录因子。总共确定了 7 个与白细胞介素-1β产生有关的 CpG 基因座,这些基因座通过遗传/meQTL 分析或因果关系推断测试存活下来。其中没有一个与各种代谢特征有显著关系。进一步的分析表明,痛风炎症而不是高尿酸血症将这些 CpG 位点与痛风联系起来。其中 6 个(,,,,,和)是痛风领域的新基因。一个()以前与痛风炎症有关。转录因子映射确定了几个潜在的转录因子,这些转录因子与差异甲基化、白细胞介素-1β产生和痛风炎症之间的联系有关。总之,这项研究揭示了几个与痛风炎症特异性相关的新基因,并为痛风炎症的生物学基础提供了更深入的认识。