Department of Biomedical Data Science, Geisel School of Medicine, Dartmouth College, Hanover, NH 03755, USA.
Nucleic Acids Res. 2020 Sep 18;48(16):e94. doi: 10.1093/nar/gkaa582.
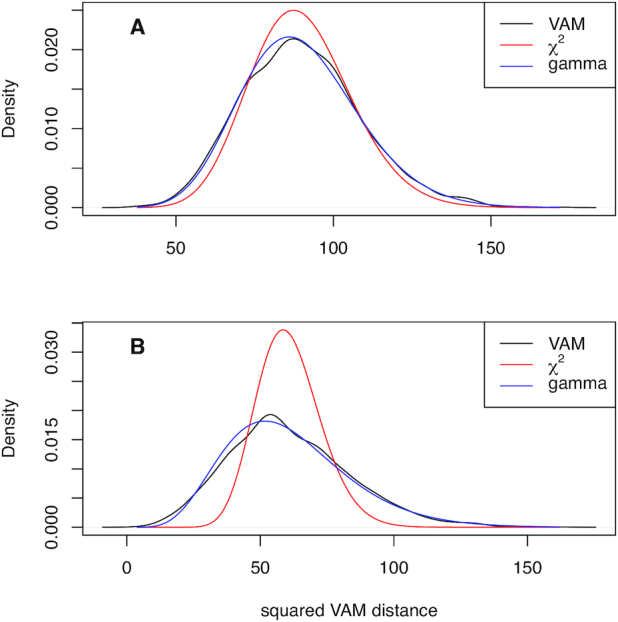
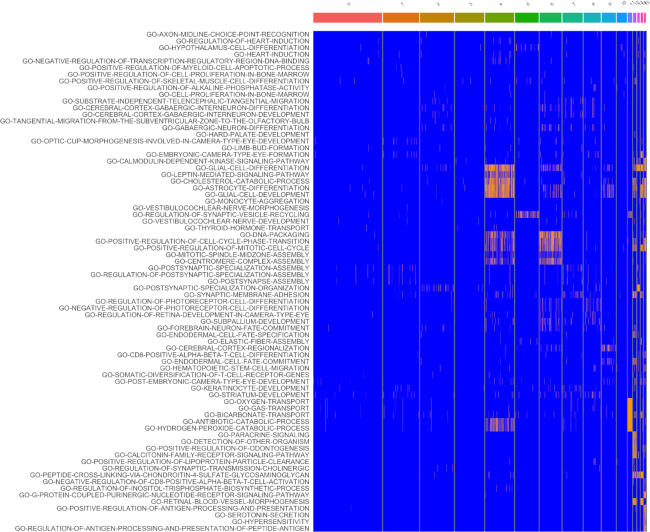
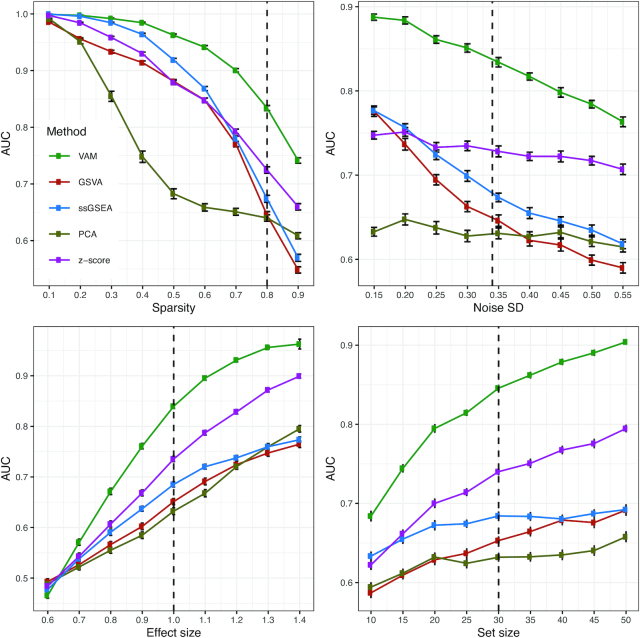
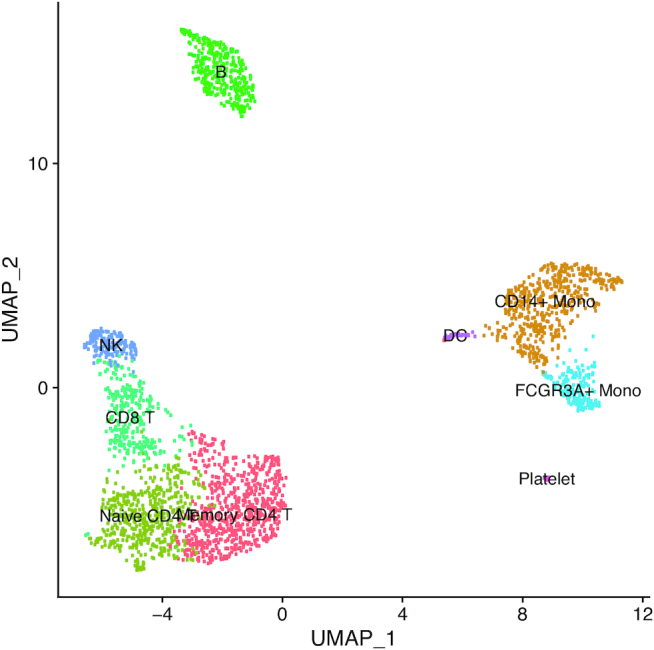
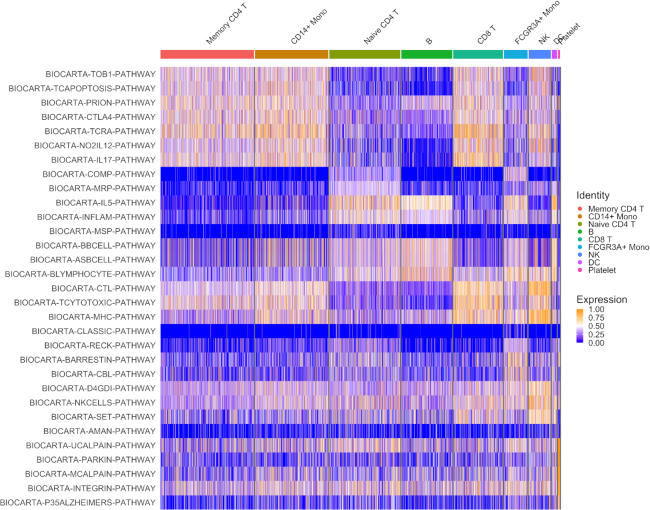
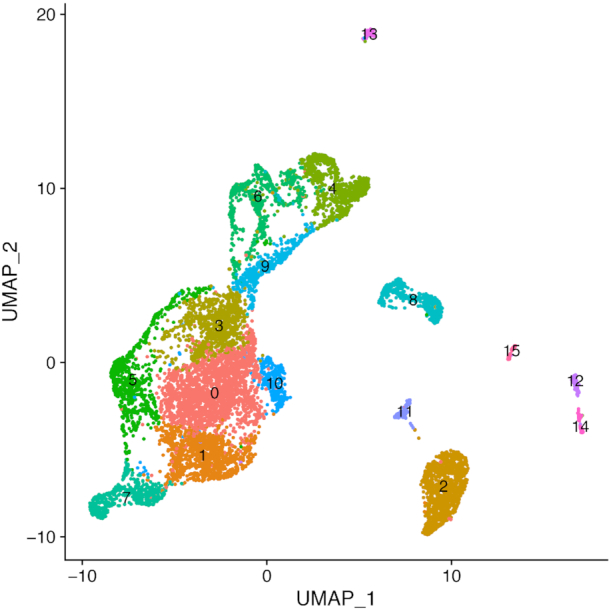
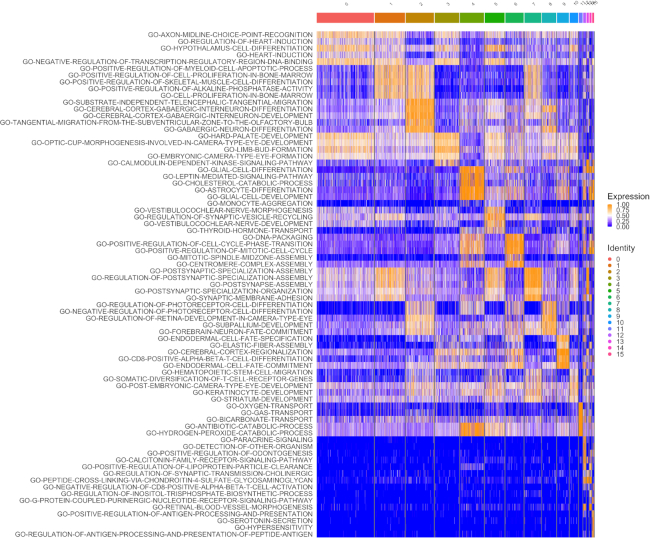
Statistical analysis of single cell RNA-sequencing (scRNA-seq) data is hindered by high levels of technical noise and inflated zero counts. One promising approach for addressing these challenges is gene set testing, or pathway analysis, which can mitigate sparsity and noise, and improve interpretation and power, by aggregating expression data to the pathway level. Unfortunately, methods optimized for bulk transcriptomics perform poorly on scRNA-seq data and progress on single cell-specific techniques has been limited. Importantly, no existing methods support cell-level gene set inference. To address this challenge, we developed a new gene set testing method, Variance-adjusted Mahalanobis (VAM), that integrates with the Seurat framework and can accommodate the technical noise, sparsity and large sample sizes characteristic of scRNA-seq data. The VAM method computes cell-specific pathway scores to transform a cell-by-gene matrix into a cell-by-pathway matrix that can be used for both data visualization and statistical enrichment analysis. Because the distribution of these scores under the null of uncorrelated technical noise has an accurate gamma approximation, both population and cell-level inference is supported. As demonstrated using simulated and real scRNA-seq data, the VAM method provides superior classification accuracy at a lower computation cost relative to existing single sample gene set testing approaches.
单细胞 RNA 测序 (scRNA-seq) 数据的统计分析受到高水平技术噪声和膨胀零计数的阻碍。一种有前途的方法是基因集测试或途径分析,通过将表达数据汇总到途径水平,可以减轻稀疏性和噪声,并提高解释和功效。不幸的是,针对批量转录组学优化的方法在 scRNA-seq 数据上表现不佳,单细胞特异性技术的进展受到限制。重要的是,没有现有的方法支持细胞水平的基因集推断。为了解决这个挑战,我们开发了一种新的基因集测试方法,即方差调整的马氏距离 (VAM),它与 Seurat 框架集成,可以适应 scRNA-seq 数据的技术噪声、稀疏性和大样本量的特点。VAM 方法计算细胞特异性途径得分,将细胞基因矩阵转换为细胞途径矩阵,可用于数据可视化和统计富集分析。由于这些得分在无相关技术噪声的零假设下的分布具有准确的伽马近似,因此支持群体和细胞水平的推断。使用模拟和真实的 scRNA-seq 数据证明,与现有的单样本基因集测试方法相比,VAM 方法在更低的计算成本下提供了更高的分类准确性。