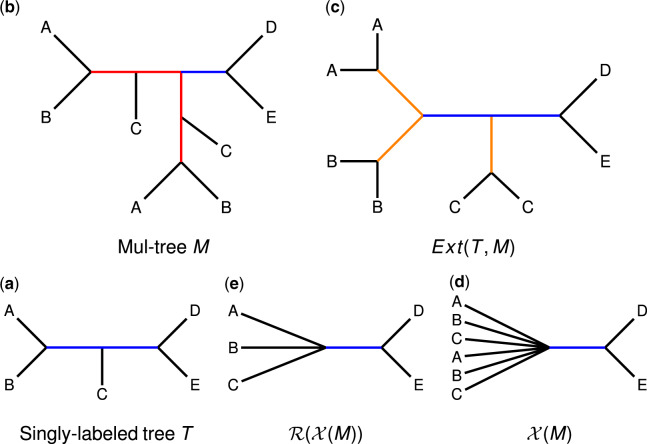
Department of Computer Science, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA.
Bioinformatics. 2020 Jul 1;36(Suppl_1):i57-i65. doi: 10.1093/bioinformatics/btaa444.
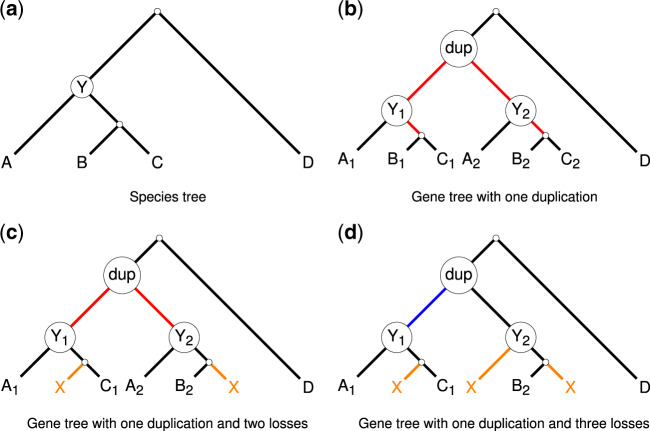
Species tree estimation is a basic part of biological research but can be challenging because of gene duplication and loss (GDL), which results in genes that can appear more than once in a given genome. All common approaches in phylogenomic studies either reduce available data or are error-prone, and thus, scalable methods that do not discard data and have high accuracy on large heterogeneous datasets are needed.
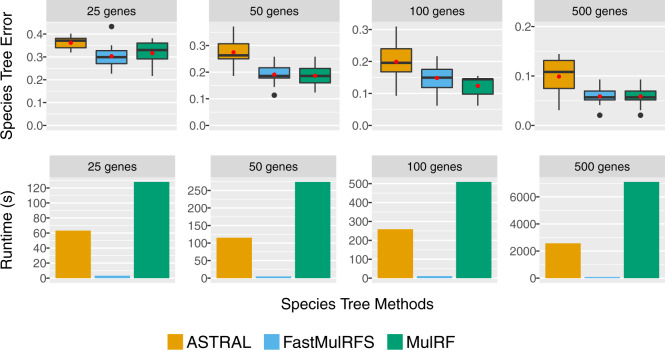
We present FastMulRFS, a polynomial-time method for estimating species trees without knowledge of orthology. We prove that FastMulRFS is statistically consistent under a generic model of GDL when adversarial GDL does not occur. Our extensive simulation study shows that FastMulRFS matches the accuracy of MulRF (which tries to solve the same optimization problem) and has better accuracy than prior methods, including ASTRAL-multi (the only method to date that has been proven statistically consistent under GDL), while being much faster than both methods.
FastMulRFS is available on Github (https://github.com/ekmolloy/fastmulrfs).
Supplementary data are available at Bioinformatics online.
物种树估计是生物研究的基本部分,但由于基因复制和丢失(GDL),这会导致给定基因组中的基因出现多次,因此具有挑战性。系统发育基因组学研究中的所有常见方法要么减少可用数据,要么容易出错,因此需要不丢弃数据且在大型异构数据集上具有高精度的可扩展方法。
我们提出了 FastMulRFS,这是一种在不知道同源性的情况下估计物种树的多项式时间方法。我们证明了在对抗性 GDL 不发生的情况下,FastMulRFS 在通用 GDL 模型下具有统计一致性。我们广泛的模拟研究表明,FastMulRFS 与 MulRF 的准确性相匹配(MulRF 试图解决相同的优化问题),并且比包括 ASTRAL-multi(迄今为止唯一在 GDL 下证明具有统计一致性的方法)在内的先前方法具有更高的准确性,同时比这两种方法都快得多。
FastMulRFS 可在 Github(https://github.com/ekmolloy/fastmulrfs)上获得。
补充数据可在 Bioinformatics 在线获得。