Delabre Mattéo, El-Mabrouk Nadia, Huber Katharina T, Lafond Manuel, Moulton Vincent, Noutahi Emmanuel, Castellanos Miguel Sautie
Département d'informatique (DIRO), Université de Montréal, Québec, Canada.
School of Computing Sciences, University of East Anglia, Norwich, UK.
Algorithms Mol Biol. 2020 May 26;15:12. doi: 10.1186/s13015-020-00171-4. eCollection 2020.
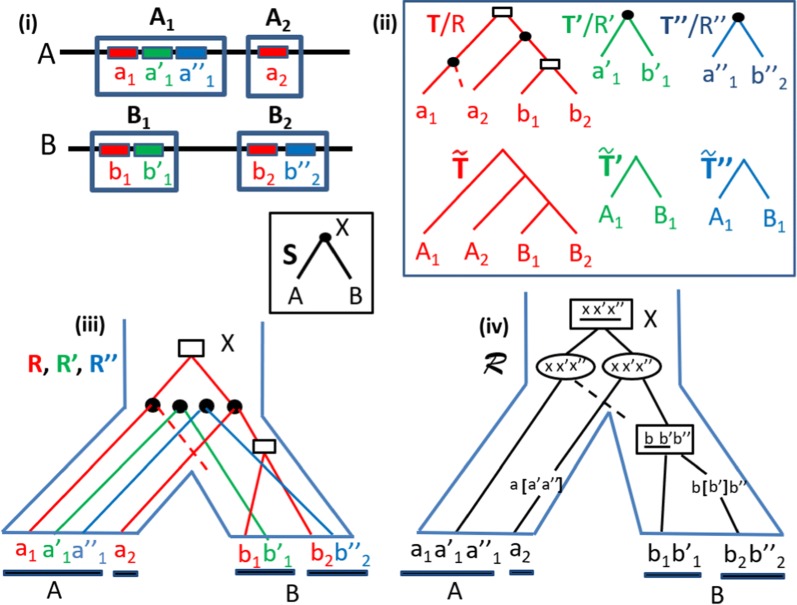
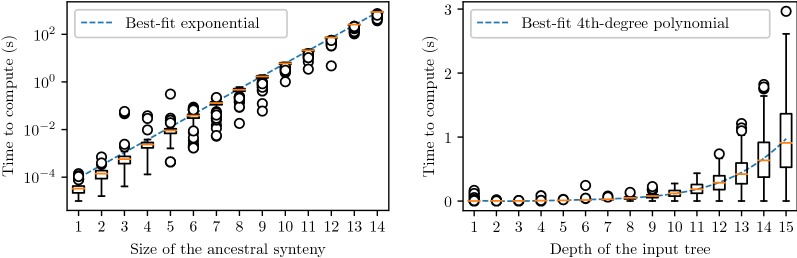
The classical gene and species tree reconciliation, used to infer the history of gene gain and loss explaining the evolution of gene families, assumes an independent evolution for each family. While this assumption is reasonable for genes that are far apart in the genome, it is not appropriate for genes grouped into syntenic blocks, which are more plausibly the result of a concerted evolution. Here, we introduce the problem which consists in inferring a history of segmental duplication and loss events (involving a set of neighboring genes) leading to a set of present-day syntenies from a single ancestral one. In other words, we extend the traditional Duplication-Loss reconciliation problem of a single gene tree, to a set of trees, accounting for segmental duplications and losses. Existency of a Super-Reconciliation depends on individual gene tree consistency. In addition, ignoring rearrangements implies that existency also depends on gene order consistency. We first show that the problem of reconstructing a most parsimonious Super-Reconciliation, if any, is NP-hard and give an exact exponential-time algorithm to solve it. Alternatively, we show that accounting for rearrangements in the evolutionary model, but still only minimizing segmental duplication and loss events, leads to an exact polynomial-time algorithm. We finally assess time efficiency of the former exponential time algorithm for the Duplication-Loss model on simulated datasets, and give a proof of concept on the opioid receptor genes.
用于推断基因家族进化过程中基因得失历史的经典基因树与物种树协调分析,假定每个基因家族独立进化。虽然这一假设对于基因组中距离较远的基因是合理的,但对于成串排列在同一条染色体上的基因则不合适,这些基因更可能是协同进化的结果。在此,我们提出一个问题,即从单一祖先染色体片段推断导致一组现今同线性区域的片段重复和缺失事件(涉及一组相邻基因)的历史。换句话说,我们将单个基因树的传统重复-缺失协调问题扩展到一组基因树,同时考虑片段重复和缺失。超级协调分析的存在性取决于各个基因树的一致性。此外,忽略重排意味着存在性还取决于基因顺序的一致性。我们首先证明,如果存在的话,重建最简约超级协调分析的问题是NP难问题,并给出一种精确的指数时间算法来解决它。另外,我们证明在进化模型中考虑重排,但仍仅最小化片段重复和缺失事件,会得到一种精确的多项式时间算法。我们最后评估了前一种指数时间算法在模拟数据集上对于重复-缺失模型的时间效率,并给出了阿片受体基因的概念验证。