Roy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6323, United States.
Biosciences Faculty, University of Heidelberg, 69120 Heidelberg, Germany.
ACS Chem Biol. 2020 Aug 21;15(8):2154-2163. doi: 10.1021/acschembio.0c00362. Epub 2020 Jul 23.
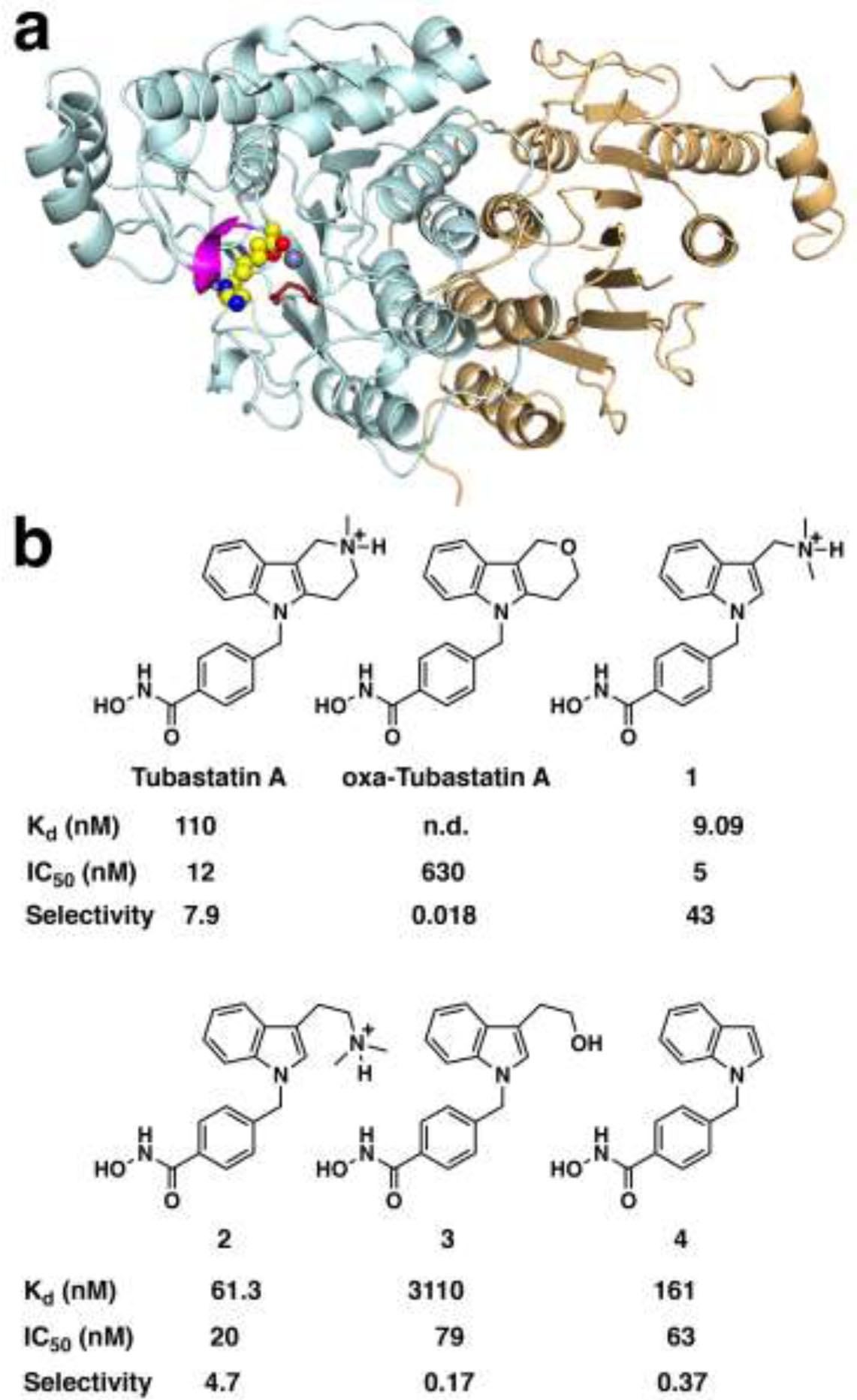
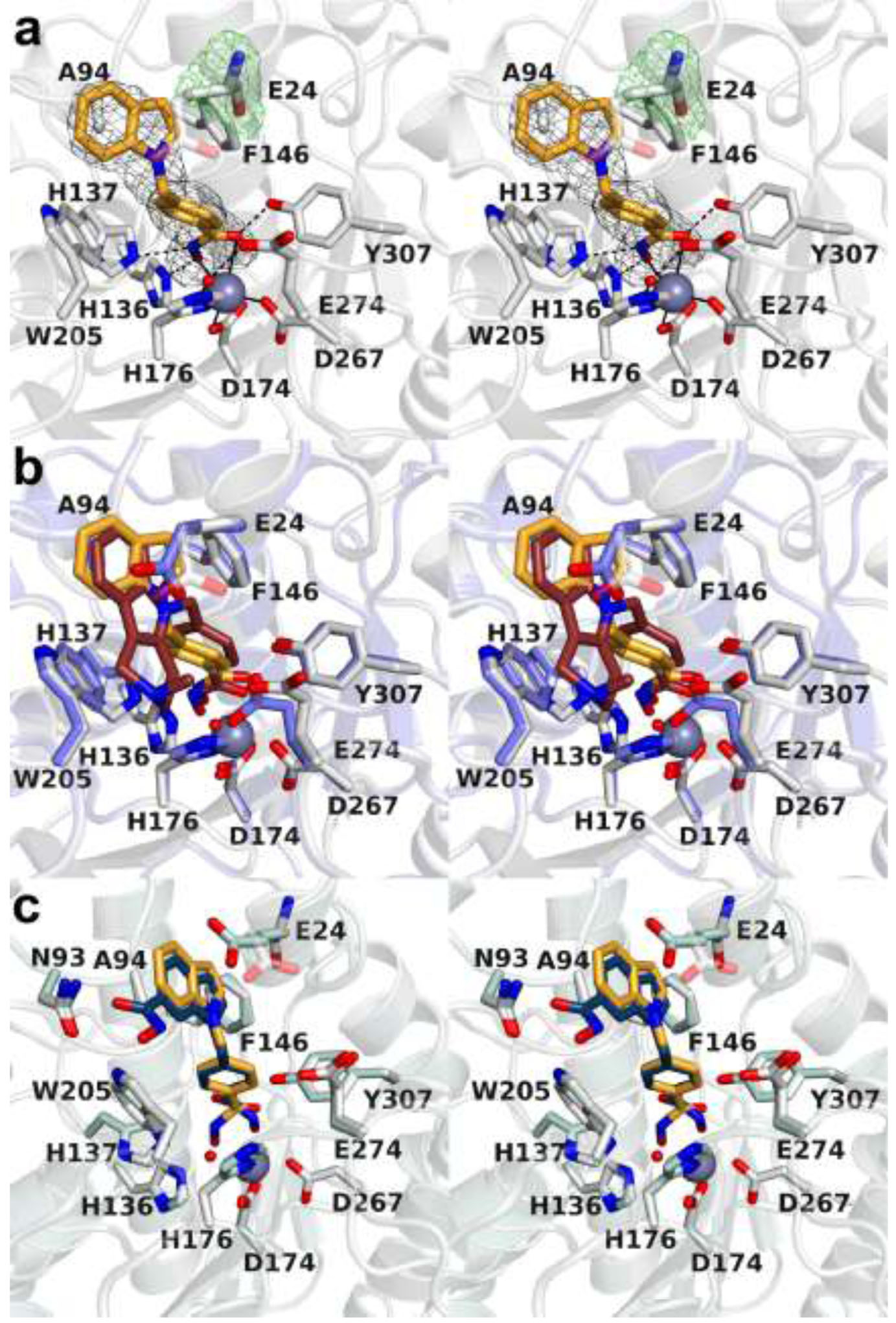
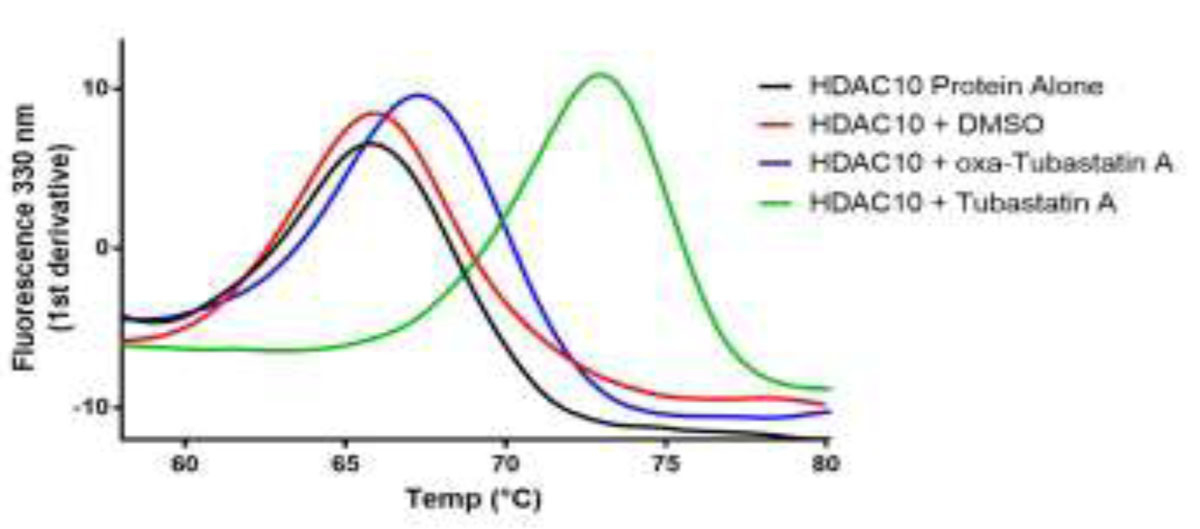
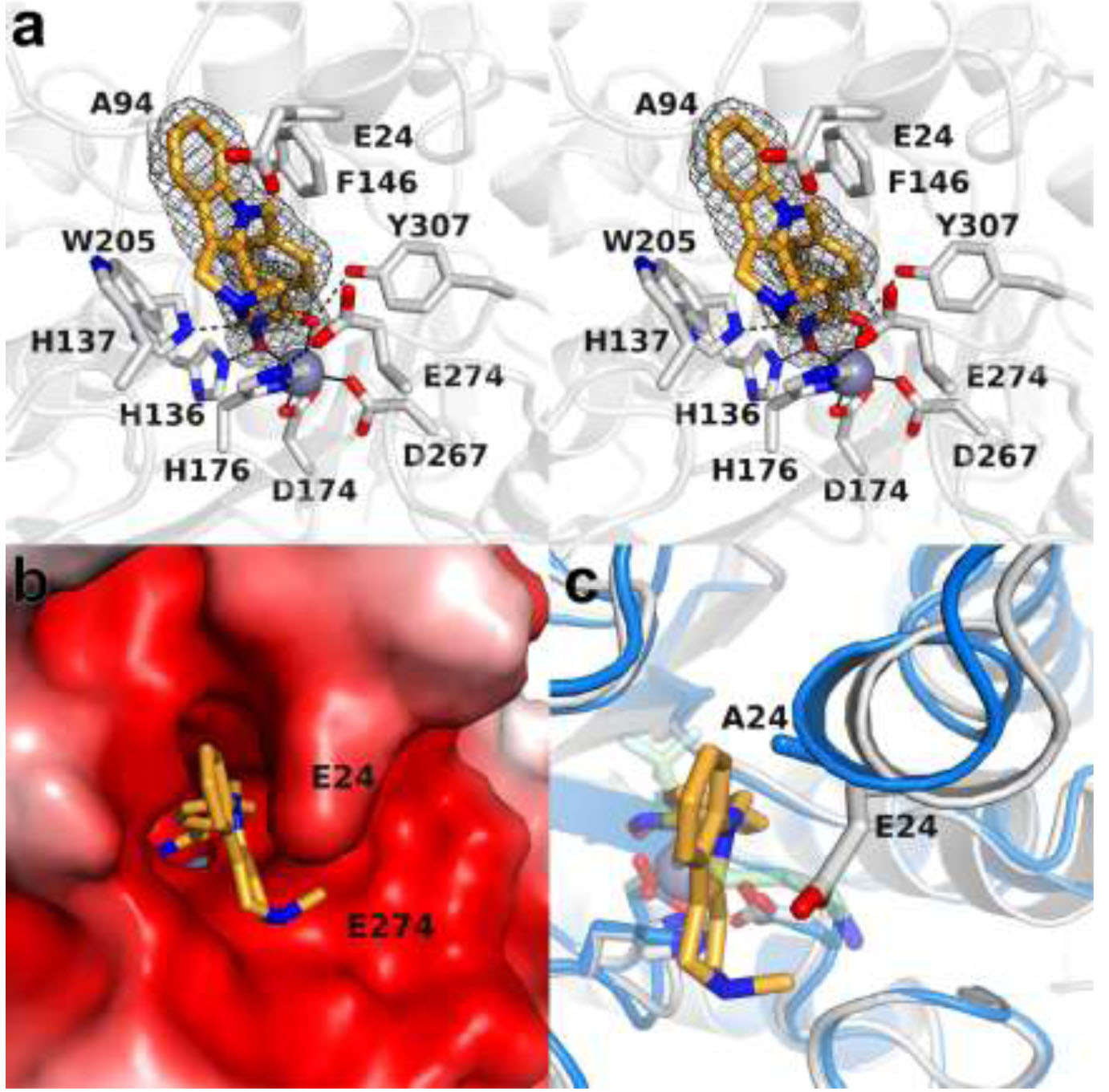
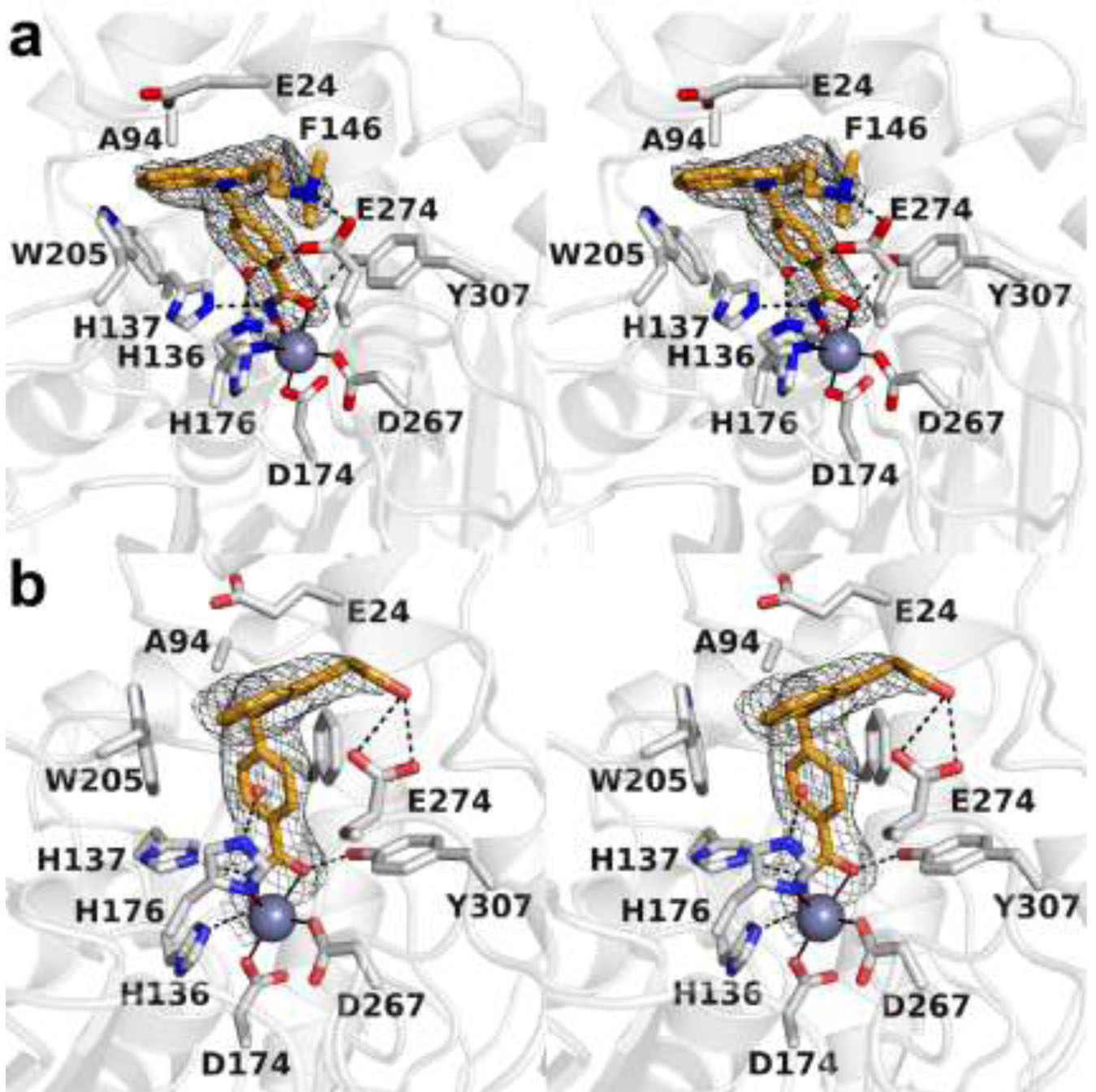
The cytosolic class IIb histone deacetylase HDAC10 is an emerging target for drug design. As an inducer of autophagy, its selective inhibition suppresses the autophagic response that otherwise attenuates the efficacy of cytotoxic cancer chemotherapy drugs. HDAC10 is a zinc-dependent polyamine deacetylase exhibiting maximal catalytic activity against -acetylspermidine. As revealed in the structure of (zebrafish) HDAC10, two conserved structural motifs direct this narrow substrate specificity: a 3 helix containing the P(E,A)CE motif that sterically constricts the active site and an electrostatic "gatekeeper," E274, that confers selectivity for cationic polyamine substrates. To accelerate drug design efforts targeting human HDAC10, we now report the preparation of "humanized" zebrafish HDAC10 in which two amino acid substitutions, A24E and D94A, yield an active site contour more similar to that of human HDAC10. X-ray crystal structures of this HDAC10 variant complexed with Tubastatin A and indole analogues bearing pendant tertiary amines reveal that inhibitors capable of hydrogen bonding with gatekeeper E274 exhibit high affinity and selectivity for HDAC10 over HDAC6 (the other class IIb isozyme). Moreover, these structures reveal that the P(E,A)CE motif helix can shift by up to 2 Å to accommodate the binding of bulky inhibitors. Thus, slender polyamine-like inhibitor structures are not exclusively required for selective, high affinity binding to HDAC10. Indeed, the flexibility of the P(E,A)CE motif helix could conceivably enable the binding of certain protein substrates.
细胞质 IIb 组蛋白去乙酰化酶 HDAC10 是药物设计的新兴靶点。作为自噬诱导剂,其选择性抑制可抑制自噬反应,否则会降低细胞毒性癌症化疗药物的疗效。HDAC10 是一种锌依赖性聚胺脱乙酰酶,对 -乙酰基 spermidine 表现出最大的催化活性。正如(斑马鱼)HDAC10 的结构所揭示的那样,两个保守的结构基序指导这种狭窄的底物特异性:包含 P(E,A)CE 基序的 3 螺旋,该基序在空间上限制了活性位点,以及静电“守门员”E274,赋予了阳离子聚胺底物的选择性。为了加速针对人 HDAC10 的药物设计工作,我们现在报告制备“人源化”斑马鱼 HDAC10,其中两个氨基酸取代 A24E 和 D94A,产生更类似于人 HDAC10 的活性位点轮廓。该 HDAC10 变体与 Tubastatin A 和带有侧接叔胺的吲哚类似物复合物的 X 射线晶体结构表明,能够与守门员 E274 形成氢键的抑制剂对 HDAC10 具有高亲和力和选择性,而对 HDAC6(另一种 IIb 同工酶)则没有。此外,这些结构表明 P(E,A)CE 基序螺旋可以移动多达 2 Å 以容纳大抑制剂的结合。因此,细长的聚胺样抑制剂结构并非选择性、高亲和力结合 HDAC10 所必需的。事实上,P(E,A)CE 基序螺旋的灵活性可以设想使某些蛋白质底物结合成为可能。