Department of Computer Science and Engineering, National Institute of Technical Teachers' Training and Research, Kolkata, West Bengal, India.
Department of Computer Science and Information Technology, Institute of Technical Education and Research, Siksha 'O' Anusandhan (Deemed to be University), Bhubaneswar, Orissa, India.
Infect Genet Evol. 2020 Nov;85:104457. doi: 10.1016/j.meegid.2020.104457. Epub 2020 Jul 11.
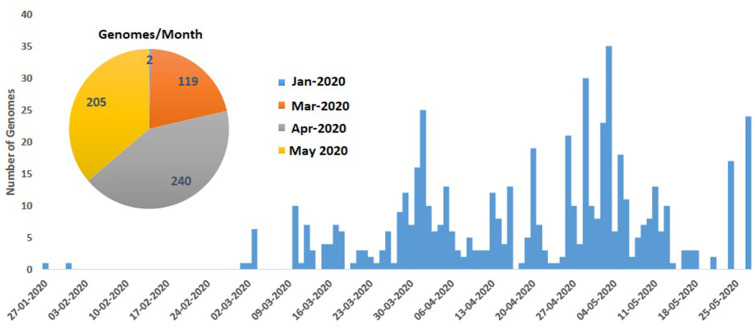
The wave of COVID-19 is a big threat to the human population. Presently, the world is going through different phases of lock down in order to stop this wave of pandemic; India being no exception. We have also started the lock down on 23rd March 2020. In this current situation, apart from social distancing only a vaccine can be the proper solution to serve the population of human being. Thus it is important for all the nations to perform the genome-wide analysis in order to identify the genetic variation in Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) so that proper vaccine can be designed. This fast motivated us to analyze publicly available 566 Indian complete or near complete SARS-CoV-2 genomes to find the mutation points as substitution, deletion and insertion. In this regard, we have performed the multiple sequence alignment in presence of reference sequence from NCBI. After the alignment, a consensus sequence is built to analyze each genome in order to identify the mutation points. As a consequence, we have found 933 substitutions, 2449 deletions and 2 insertions, in total 3384 unique mutation points, in 566 genomes across 29.9 K bp. Further, it has been classified into three groups as 100 clusters of mutations (mostly deletions), 1609 point mutations as substitution, deletion and insertion and 64 SNPs. These outcomes are visualized using BioCircos and bar plots as well as plotting entropy value of each genomic location. Moreover, phylogenetic analysis has also been performed to see the evolution of SARS-CoV-2 virus in India. It also shows the wide variation in tree which indeed vivid in genomic analysis. Finally, these SNPs can be the useful target for virus classification, designing and defining the effective dose of vaccine for the heterogeneous population.
新冠疫情浪潮对人类构成了巨大威胁。目前,全球各国正处于不同阶段的封锁之中,以阻止这一波大流行;印度也不例外。我们已于 2020 年 3 月 23 日开始实施封锁。在这种情况下,除了保持社交距离,疫苗是应对人类人口的唯一有效方法。因此,所有国家都有必要进行全基因组分析,以确定严重急性呼吸系统综合征冠状病毒 2 型(SARS-CoV-2)的遗传变异,从而设计出合适的疫苗。这一快速发展促使我们分析了公开的 566 个印度完整或近乎完整的 SARS-CoV-2 基因组,以发现突变点,如替换、缺失和插入。在这方面,我们在参考序列来自 NCBI 的情况下进行了多次序列比对。在比对之后,构建了一个共识序列来分析每个基因组,以识别突变点。结果,我们在 566 个基因组中共发现了 3384 个独特的突变点,包括 933 个替换、2449 个缺失和 2 个插入,跨越了 29.9 Kbp。进一步将其分为三组:100 个突变簇(主要是缺失)、1609 个点突变(替换、缺失和插入)和 64 个单核苷酸多态性。这些结果使用 BioCircos 和条形图以及每个基因组位置的熵值图进行可视化。此外,还进行了系统发育分析,以观察 SARS-CoV-2 病毒在印度的进化情况。这也表明了树的广泛变异,这在基因组分析中确实生动。最后,这些单核苷酸多态性可以作为病毒分类、设计和确定异质人群有效疫苗剂量的有用目标。