Radiation & Cancer Biology Laboratory, Radiation Oncology Center, Chongqing Key Laboratory of Translational Research for Cancer Metastasis and Individualized Treatment, Chongqing University Cancer Hospital & Chongqing Cancer, Institute & Chongqing Cancer Hospital, Chongqing 400030, China.
The Department of Internal Medicine, Chongqing University Cancer Hospital & Chongqing Cancer, Institute & Chongqing Cancer Hospital, Chongqing 400030, China.
Comput Intell Neurosci. 2021 Dec 31;2021:9444194. doi: 10.1155/2021/9444194. eCollection 2021.
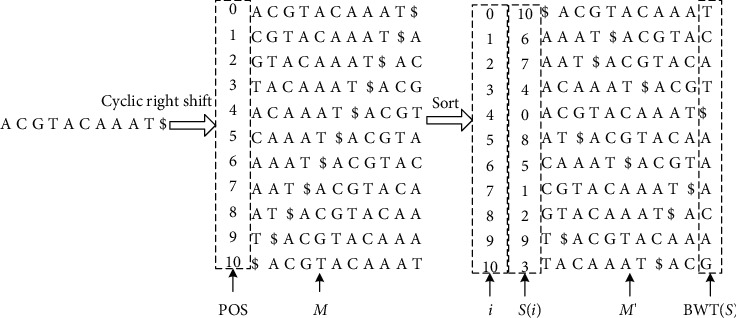
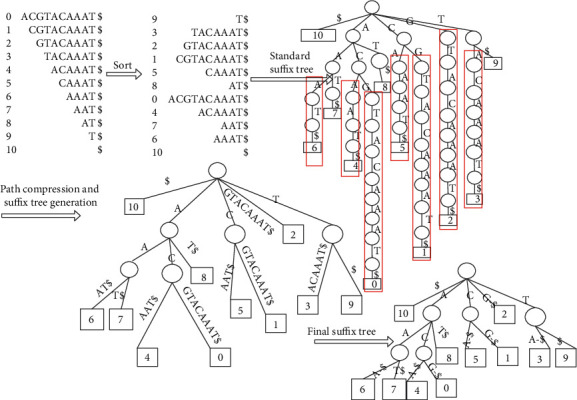
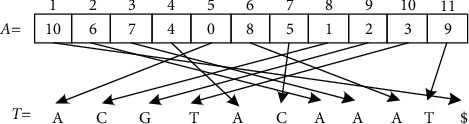
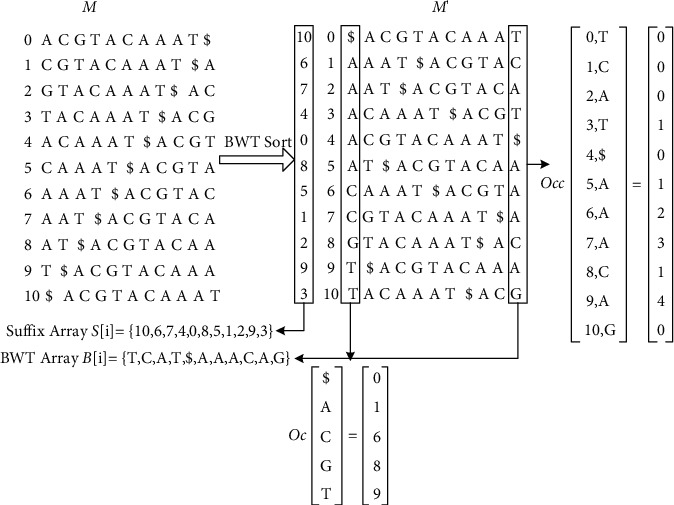
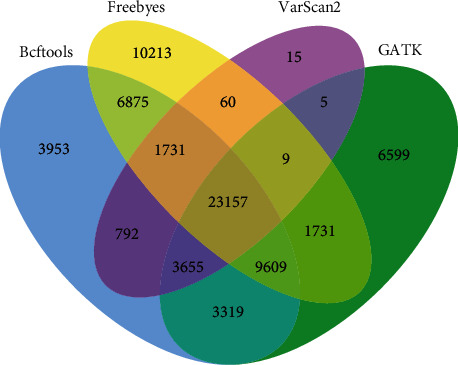
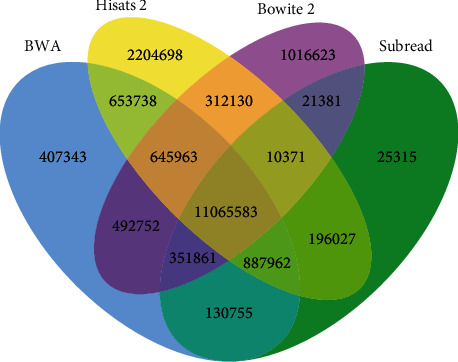
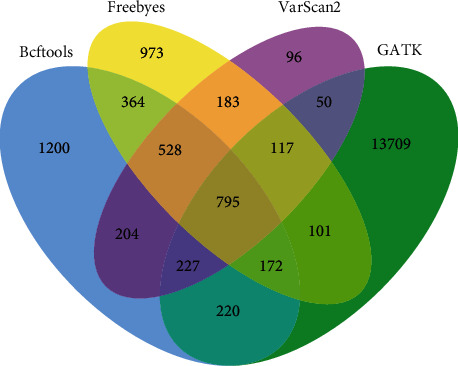
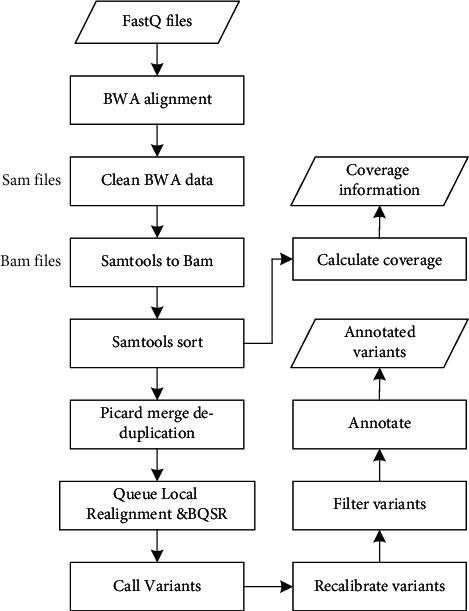
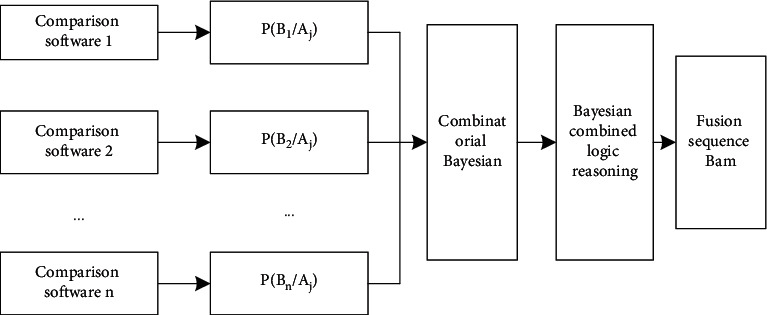
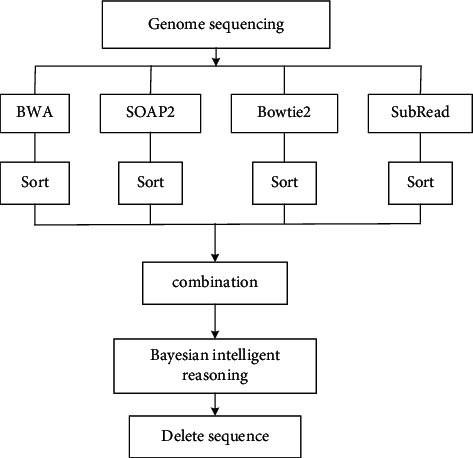
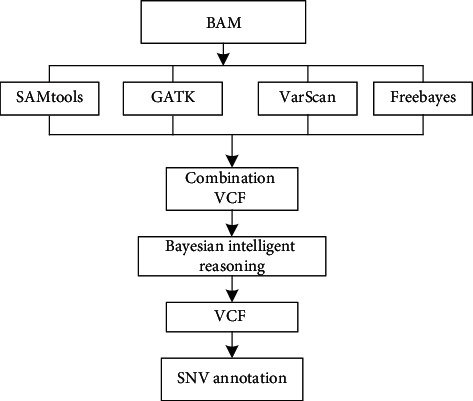
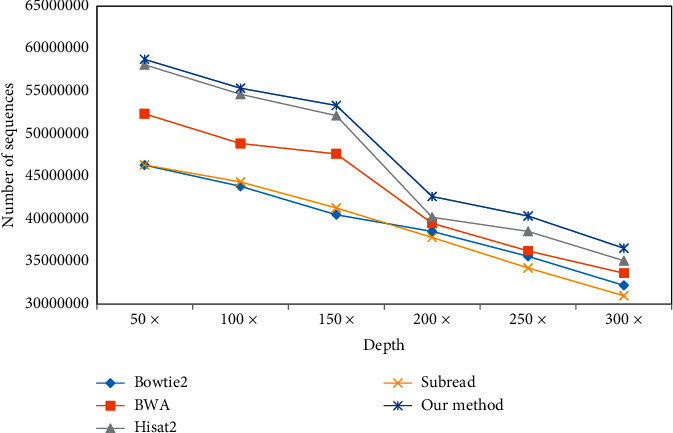
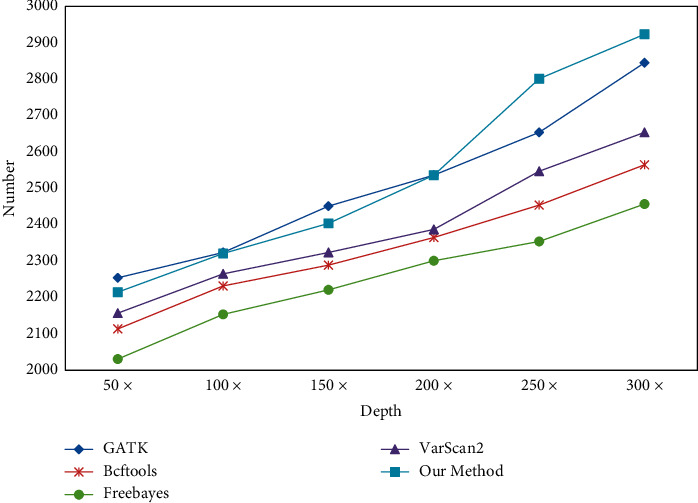
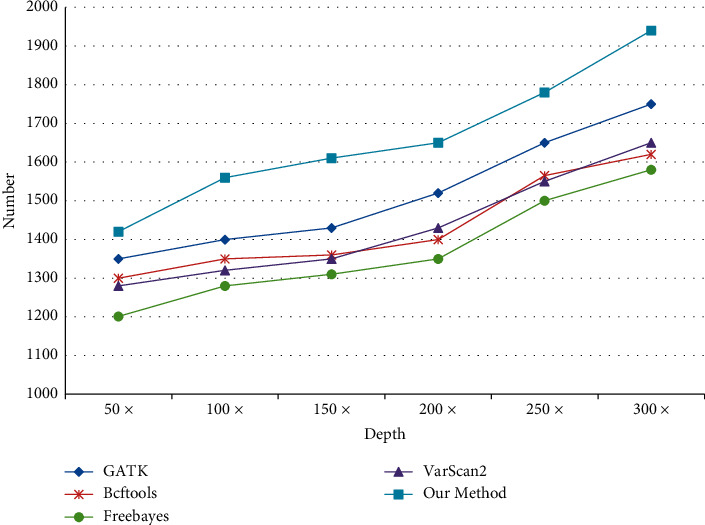
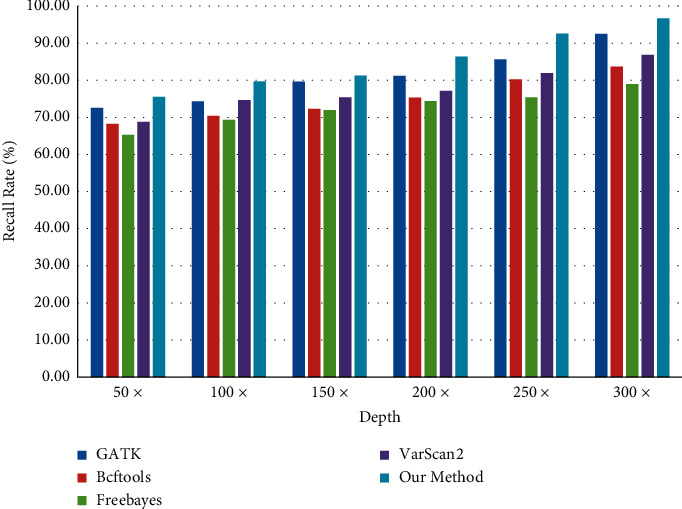
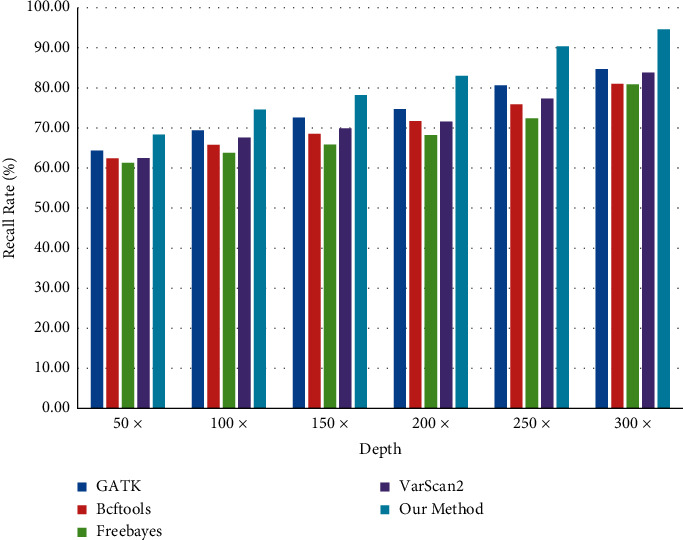
With the rapid development of DNA high-throughput testing technology, there is a high correlation between DNA sequence variation and human diseases, and detecting whether there is variation in DNA sequence has become a hot research topic at present. DNA sequence variation is relatively rare, and the establishment of DNA sequence sparse matrix, which can quickly detect and reason fusion variation point, has become an important work of tumor gene testing. Because there are differences between the current comparison software and mutation detection software in detecting the same sample, there are errors between the results of derivative sequence comparison and the detection of mutation. In this paper, SNP and InDel detection methods based on machine learning and sparse matrix detection are proposed, and VarScan 2, Genome Analysis Toolkit (GATK), BCFtools, and FreeBayes are compared. In the research of SNP and InDel detection with intelligent reasoning, the experimental results show that the detection accuracy and recall rate are better when the depth is increasing. The reasoning fusion method proposed in this paper has certain advantages in comparison effect and discovery in SNP and InDel and has good effect on swelling and pain gene detection.
随着 DNA 高通量检测技术的飞速发展,DNA 序列变异与人类疾病之间存在高度相关性,检测 DNA 序列是否存在变异已成为当前的热门研究课题。DNA 序列变异相对较少,建立能够快速检测和推理融合变异点的 DNA 序列稀疏矩阵已成为肿瘤基因检测的重要工作。由于当前比较软件和突变检测软件在检测同一样本时存在差异,因此衍生序列比较结果与突变检测之间存在误差。本文提出了基于机器学习和稀疏矩阵检测的 SNP 和 InDel 检测方法,并对 VarScan 2、基因组分析工具包(GATK)、BCFtools 和 FreeBayes 进行了比较。在智能推理 SNP 和 InDel 检测的研究中,实验结果表明,随着深度的增加,检测的准确性和召回率更好。本文提出的推理融合方法在 SNP 和 InDel 的比较效果和发现方面具有一定的优势,对肿胀和疼痛基因检测具有良好的效果。