Natural Product Informatics Research Center, KIST Gangneung Institute of Natural Products, Gangneung, 25451, Republic of Korea.
Department of Bioinformatics and Life Science, Soongsil University, Seoul, 06978, Republic of Korea.
BMC Bioinformatics. 2020 Jul 14;21(1):309. doi: 10.1186/s12859-020-03643-x.
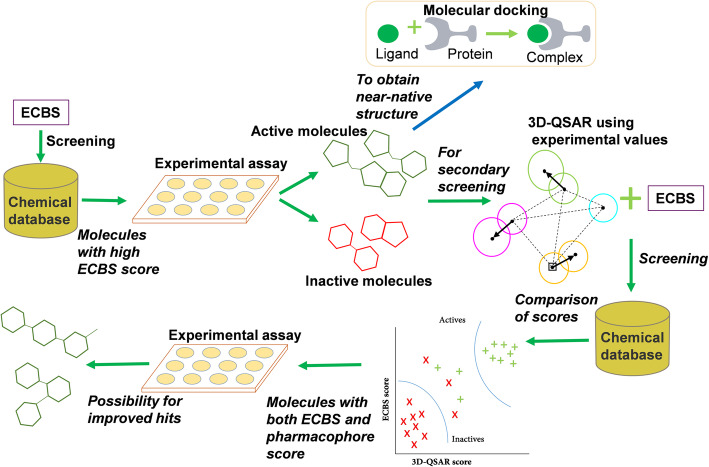
Despite continued efforts using chemical similarity methods in virtual screening, currently developed approaches suffer from time-consuming multistep procedures and low success rates. We recently developed a machine learning-based chemical binding similarity model considering common structural features from molecules binding to the same, or evolutionarily related targets. The chemical binding similarity measures the resemblance of chemical compounds in terms of binding site similarity to better describe functional similarities that arise from target binding. In this study, we have shown how the chemical binding similarity could be used in virtual screening together with the conventional structure-based methods.
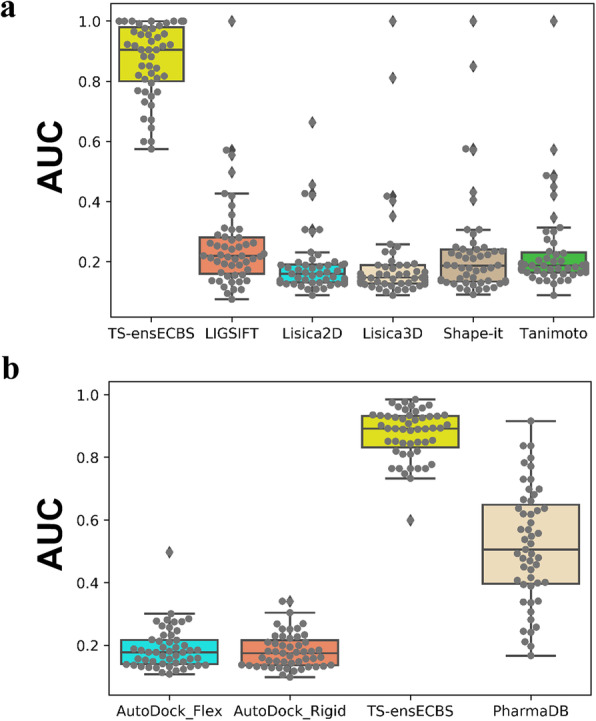
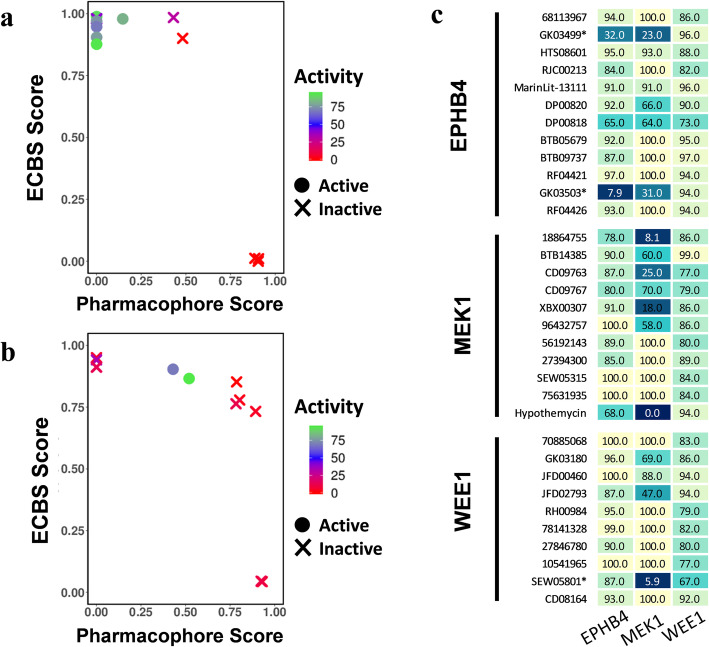
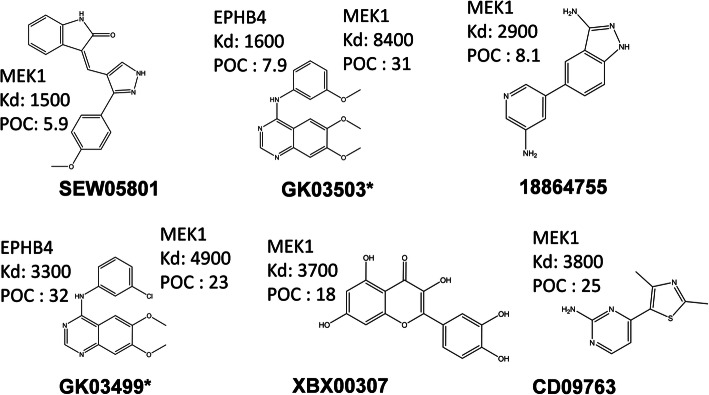
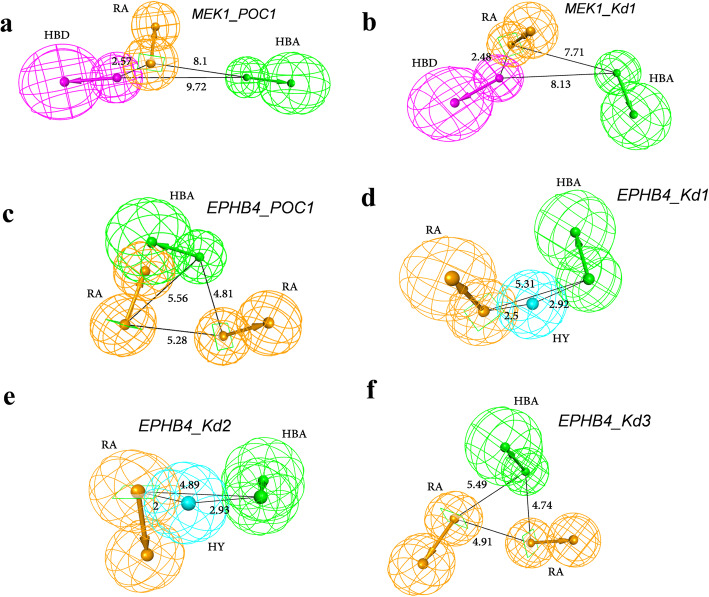
The chemical binding similarity, receptor-based pharmacophore, chemical structure similarity, and molecular docking methods were evaluated to identify an effective virtual screening procedure for desired target proteins. When we tested the chemical binding similarity method with test sets of 51 kinases, it outperformed the traditional structural similarity-based methods as well as structure-based methods, such as molecular docking and receptor-based pharmacophore modeling, in terms of finding active compounds. We further validated the results by performing virtual screening (using the chemical binding similarity and receptor-based pharmacophore methods) against a completely blind dataset for mitogen-activated protein kinase kinase 1 (MEK1), ephrin type-B receptor 4 (EPHB4) and wee1-like protein kinase (WEE1). The in vitro kinase binding assay confirmed that 6 out of 13 (46.2%) for MEK1 and 2 out of 12 (16.7%) for EPHB4 were newly identified only by the chemical binding similarity model.
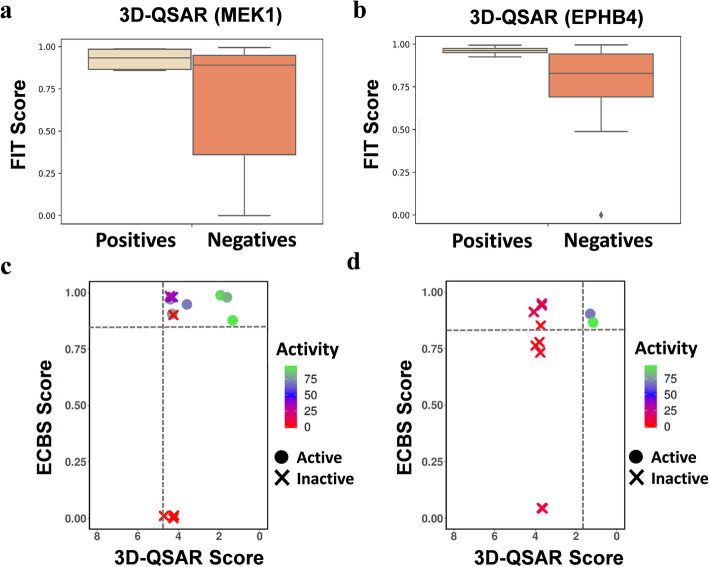
We report that the virtual screening results could further be improved by combining the chemical binding similarity model with 3D-QSAR pharmacophore and molecular docking models. Not only the new inhibitors are identified in this study, but also many of the identified molecules have low structural similarity scores against already reported inhibitors and that show the revelation of novel scaffolds.
尽管在虚拟筛选中使用化学相似性方法的努力仍在继续,但目前开发的方法存在耗时的多步骤程序和低成功率的问题。我们最近开发了一种基于机器学习的化学结合相似性模型,该模型考虑了与相同或进化相关靶标结合的分子的常见结构特征。化学结合相似性根据结合部位的相似性来衡量化合物的相似性,以更好地描述由于靶标结合而产生的功能相似性。在这项研究中,我们展示了如何将化学结合相似性与传统的基于结构的方法一起用于虚拟筛选。
评估了化学结合相似性、基于受体的药效基团、化学结构相似性和分子对接方法,以确定针对所需靶蛋白的有效虚拟筛选程序。当我们使用 51 种激酶的测试集测试化学结合相似性方法时,它在发现活性化合物方面优于传统的基于结构的相似性方法以及基于结构的方法,例如分子对接和基于受体的药效基团建模。我们通过针对丝裂原活化蛋白激酶激酶 1(MEK1)、表皮生长因子受体 B4(EPHB4)和 wee1 样蛋白激酶(WEE1)的完全盲数据集进行虚拟筛选(使用化学结合相似性和基于受体的药效基团方法)进一步验证了结果。体外激酶结合测定证实,MEK1 中有 6 种(46.2%)和 EPHB4 中有 2 种(16.7%)是仅通过化学结合相似性模型新发现的抑制剂。
我们报告说,通过将化学结合相似性模型与 3D-QSAR 药效基团和分子对接模型相结合,虚拟筛选结果可以进一步得到改善。本研究不仅发现了新的抑制剂,而且许多鉴定出的分子与已报道的抑制剂的结构相似性评分较低,这表明了新骨架的揭示。