Sugiyama Yohei, Shimura Masaru, Ogawa-Tominaga Minako, Ebihara Tomohiro, Kinouchi Yoshina, Isozaki Keitaro, Matsuhashi Tetsuro, Tajika Makiko, Fushimi Takuya, Ichimoto Keiko, Matsunaga Ayako, Ishida Tomoki, Mizutani Kayo, Tsuruoka Tomoko, Murayama Kei
Center for Medical Genetics, Chiba Children's Hospital, 579-1 Heta-cho, Midori-ku, Chiba city, Chiba 266-0007, Japan.
Department of Metabolism, Chiba Children's Hospital, 579-1 Heta-cho, Midori-ku, Chiba city, Chiba 266-0007, Japan.
Mol Genet Metab Rep. 2020 Jul 8;24:100622. doi: 10.1016/j.ymgmr.2020.100622. eCollection 2020 Sep.
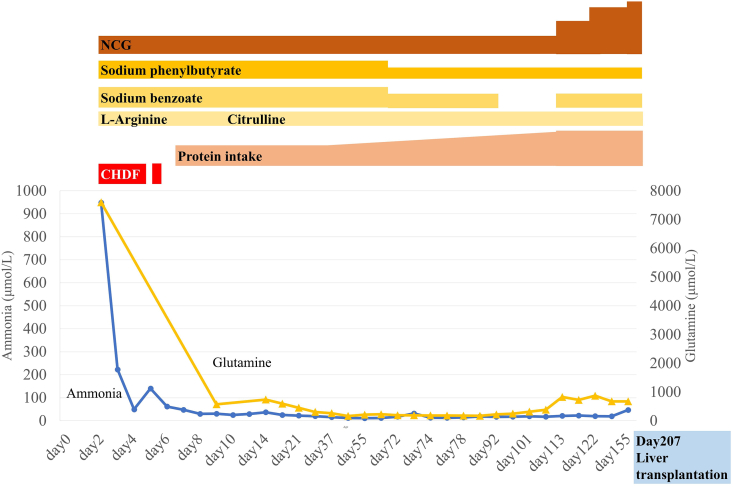
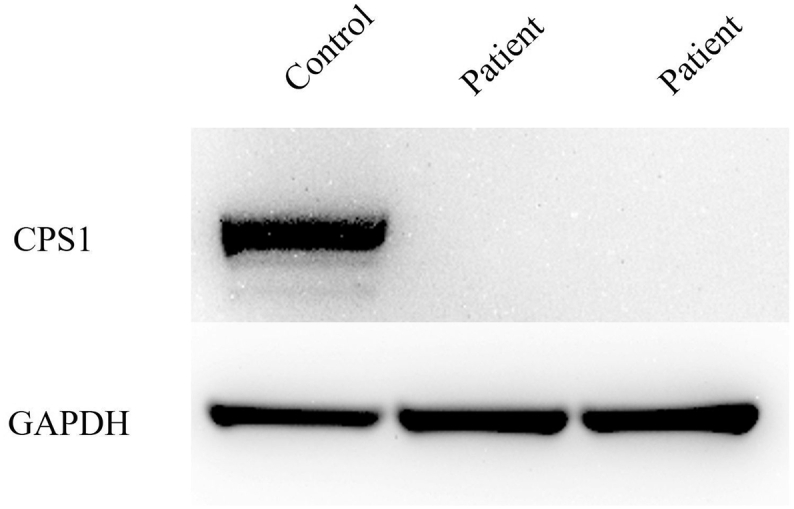
The detoxification of ammonia to urea requires a functional hepatic urea cycle, which consists of six enzymes and two mitochondrial membrane transporters. The initial step of the urea cycle is catalyzed by carbamyl phosphate synthetase 1 (CPS1). CPS1 deficiency (CPS1D) is a rare autosomal recessive disorder. -Carbamylglutamate (NCG), a deacylase-resistant analogue of -acetylglutamate, can activate CPS1. We describe the therapeutic course of a patient suffering from neonatal onset CPS1D with compound heterozygosity for the c.2359C > T (p.Arg787*) and c.3559G > T (p.Val1187Phe) variants in , treated with NCG. She presented with hyperammonemia, which reached 944 μmol/L at the age of 2 days. The ammonia concentration decreased after treatment with continuous hemodiafiltration, NCG, sodium benzoate, sodium phenylbutyrate, L-arginine, vitamin cocktail (vitamin B1, vitamin B12, vitamin C, vitamin E, biotin), l-carnitine, coenzyme Q10, and parenteral nutrition. Her ammonia and glutamine levels remained low; thus, protein intake was increased to 1.2 g/kg/day. Furthermore, the amount of sodium benzoate and sodium phenylbutyrate were reduced. She remained metabolically stable and experienced no metabolic crisis following treatment with oral NCG, sodium benzoate, sodium phenylbutyrate, citrulline, vitamin cocktail, l-carnitine, and coenzyme Q10 until she underwent liver transplantation at 207 days of age. She had no neurological complications at the age of 15 months. Ammonia and glutamine levels of the patient were successfully maintained at a low level via NCG treatment with increased protein intake, which led to normal neurological development. Thus, undiagnosed urea cycle disorders should be treated rapidly with acute therapy including NCG, which should be maintained until a genetic diagnosis is reached. It is essential to prevent metabolic crises in patients with CPS1D until liver transplantation to improve their prognoses.
氨解毒生成尿素需要一个功能正常的肝脏尿素循环,该循环由六种酶和两种线粒体膜转运蛋白组成。尿素循环的第一步由氨甲酰磷酸合成酶1(CPS1)催化。CPS1缺乏症(CPS1D)是一种罕见的常染色体隐性疾病。N-氨甲酰谷氨酸(NCG)是N-乙酰谷氨酸的一种抗脱酰酶类似物,可激活CPS1。我们描述了一名患有新生儿期起病的CPS1D患者的治疗过程,该患者的CPS1基因存在c.2359C>T(p.Arg787*)和c.3559G>T(p.Val1187Phe)复合杂合变异,接受了NCG治疗。她表现为高氨血症,2天时氨水平达到944μmol/L。经过持续血液透析滤过、NCG、苯甲酸钠、苯丁酸钠、L-精氨酸、维生素合剂(维生素B1、维生素B12、维生素C、维生素E、生物素)、左旋肉碱、辅酶Q10和肠外营养治疗后,氨浓度下降。她的氨和谷氨酰胺水平保持较低;因此,蛋白质摄入量增加到1.2g/kg/天。此外,苯甲酸钠和苯丁酸钠的用量减少。在接受口服NCG、苯甲酸钠、苯丁酸钠、瓜氨酸、维生素合剂、左旋肉碱和辅酶Q10治疗后,她一直保持代谢稳定,直到207天时接受肝移植,期间未发生代谢危机。15个月大时她没有神经并发症。通过增加蛋白质摄入量并使用NCG治疗,患者的氨和谷氨酰胺水平成功维持在低水平,从而实现了正常的神经发育。因此,未确诊的尿素循环障碍应迅速采用包括NCG在内的急性治疗方法进行治疗,并且应持续治疗直至做出基因诊断。在进行肝移植之前,预防CPS1D患者发生代谢危机对于改善其预后至关重要。