Department of Biomedical Engineering, University of Southern California, Los Angeles, CA, USA.
Mork Family Department of Chemical Engineering and Materials Science, University of Southern California, Los Angeles, CA, USA.
Cell Commun Signal. 2020 Jul 17;18(1):114. doi: 10.1186/s12964-020-00595-w.
Angiogenesis plays an important role in the survival of tissues, as blood vessels provide oxygen and nutrients required by the resident cells. Thus, targeting angiogenesis is a prominent strategy in many different settings, including both tissue engineering and cancer treatment. However, not all of the approaches that modulate angiogenesis lead to successful outcomes. Angiogenesis-based therapies primarily target pro-angiogenic factors such as vascular endothelial growth factor-A (VEGF) or fibroblast growth factor (FGF) in isolation, and there is a limited understanding of how these promoters combine together to stimulate angiogenesis. Targeting one pathway could be insufficient, as alternative pathways may compensate, diminishing the overall effect of the treatment strategy.
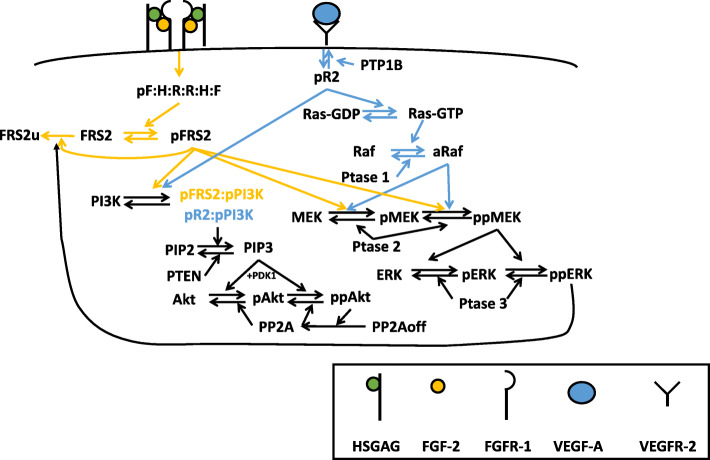
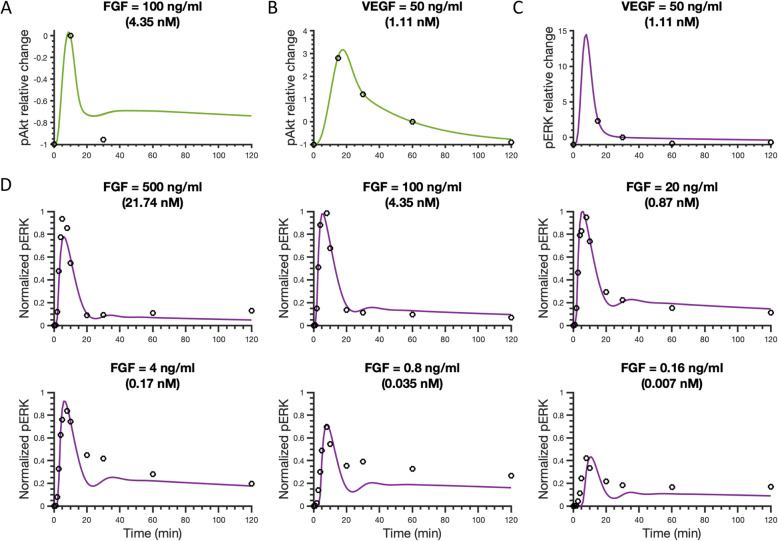
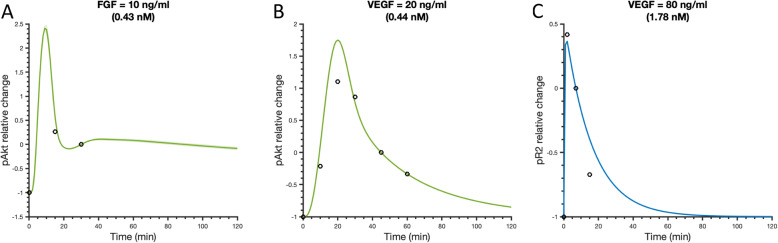
To gain mechanistic insight and identify novel therapeutic strategies, we have developed a detailed mathematical model to quantitatively characterize the crosstalk of FGF and VEGF intracellular signaling. The model focuses on FGF- and VEGF-induced mitogen-activated protein kinase (MAPK) signaling to promote cell proliferation and the phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) pathway, which promotes cell survival and migration. We fit the model to published experimental datasets that measure phosphorylated extracellular regulated kinase (pERK) and Akt (pAkt) upon FGF or VEGF stimulation. We validate the model with separate sets of data.
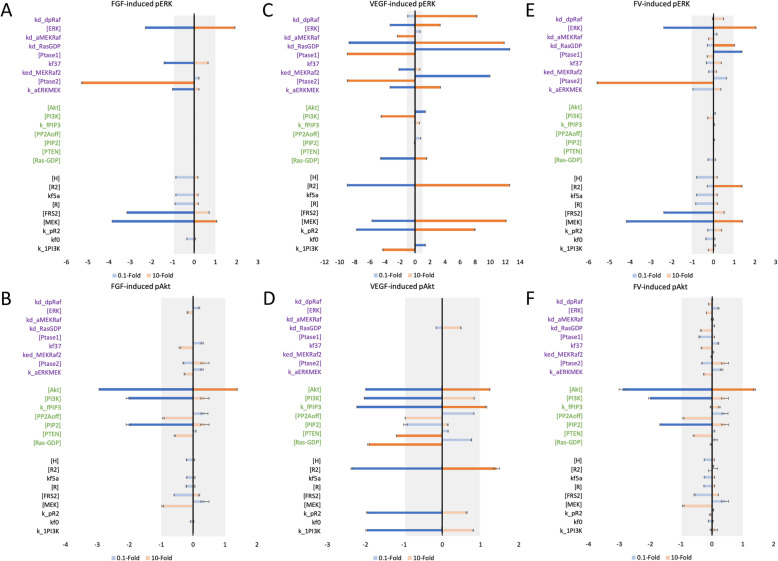
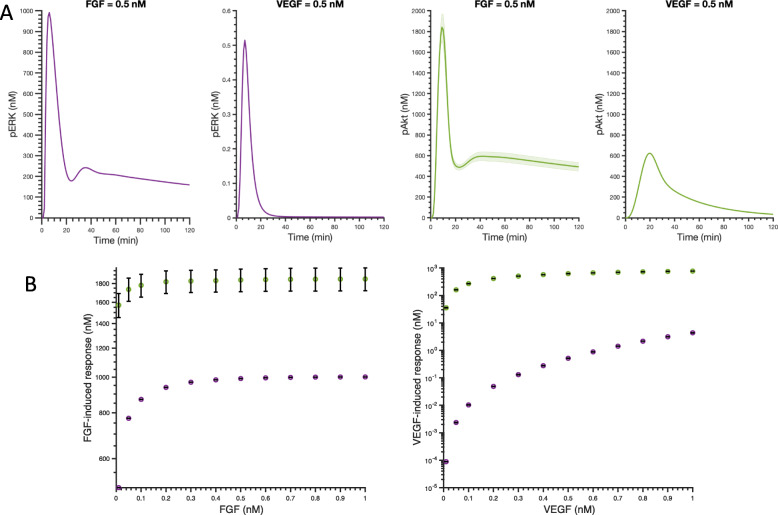
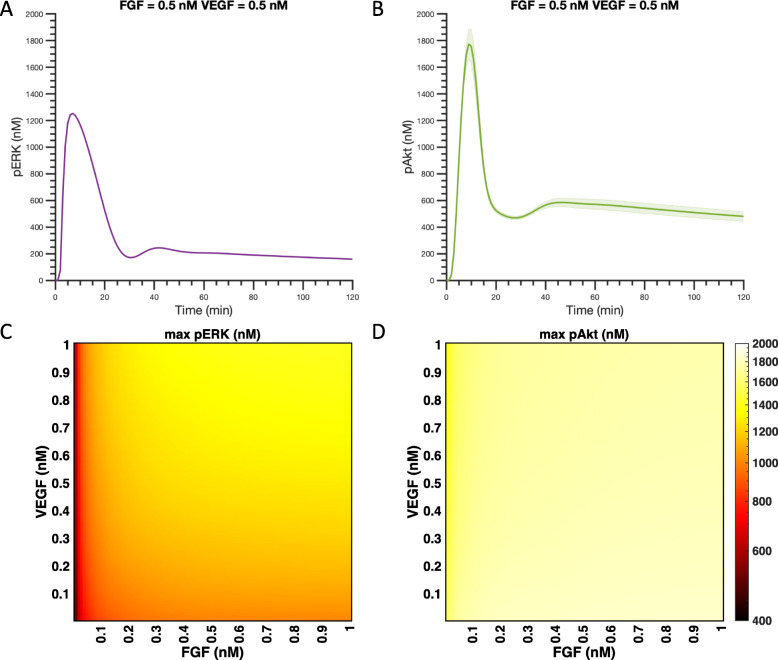
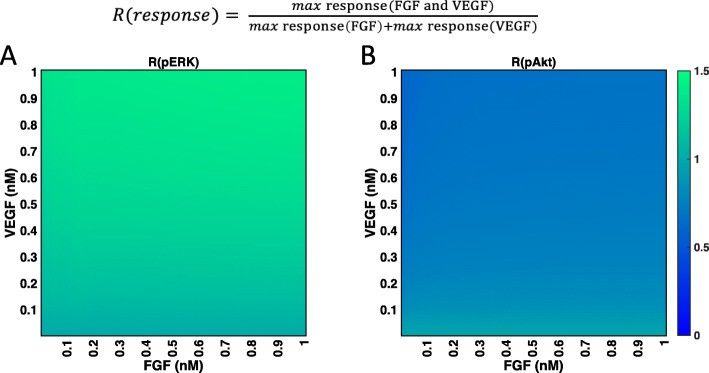
We apply the trained and validated mathematical model to characterize the dynamics of pERK and pAkt in response to the mono- and co-stimulation by FGF and VEGF. The model predicts that for certain ranges of ligand concentrations, the maximum pERK level is more responsive to changes in ligand concentration compared to the maximum pAkt level. Also, the combination of FGF and VEGF indicates a greater effect in increasing the maximum pERK compared to the summation of individual effects, which is not seen for maximum pAkt levels. In addition, our model identifies the influential species and kinetic parameters that specifically modulate the pERK and pAkt responses, which represent potential targets for angiogenesis-based therapies.
Overall, the model predicts the combination effects of FGF and VEGF stimulation on ERK and Akt quantitatively and provides a framework to mechanistically explain experimental results and guide experimental design. Thus, this model can be utilized to study the effects of pro- and anti-angiogenic therapies that particularly target ERK and/or Akt activation upon stimulation with FGF and VEGF. Video Abstract.
血管生成在组织存活中起着重要作用,因为血管为驻留细胞提供所需的氧气和营养物质。因此,靶向血管生成是许多不同环境(包括组织工程和癌症治疗)中的突出策略。然而,并非所有调节血管生成的方法都能带来成功的结果。基于血管生成的治疗主要针对血管内皮生长因子-A(VEGF)或成纤维细胞生长因子(FGF)等促血管生成因子,而对这些促进剂如何共同刺激血管生成的了解有限。靶向一种途径可能是不够的,因为替代途径可能会代偿,从而降低治疗策略的整体效果。
为了深入了解机制并确定新的治疗策略,我们开发了一个详细的数学模型,以定量描述 FGF 和 VEGF 细胞内信号的串扰。该模型侧重于 FGF 和 VEGF 诱导的丝裂原活化蛋白激酶(MAPK)信号转导,以促进细胞增殖和磷酸肌醇 3-激酶/蛋白激酶 B(PI3K/Akt)途径,促进细胞存活和迁移。我们将模型拟合到测量 FGF 或 VEGF 刺激后磷酸化细胞外调节激酶(pERK)和 Akt(pAkt)的已发表实验数据集。我们使用单独的数据集验证模型。
我们应用经过训练和验证的数学模型来描述 FGF 和 VEGF 单刺激和共刺激时 pERK 和 pAkt 的动力学。该模型预测,对于某些配体浓度范围,最大 pERK 水平对配体浓度变化的响应比对最大 pAkt 水平的响应更为敏感。此外,与单独作用相比,FGF 和 VEGF 的组合在增加最大 pERK 方面显示出更大的效果,而在最大 pAkt 水平方面则没有这种情况。此外,我们的模型确定了特定调节 pERK 和 pAkt 反应的有影响力的物种和动力学参数,这些参数可能成为基于血管生成的治疗的潜在靶点。
总的来说,该模型定量预测了 FGF 和 VEGF 刺激对 ERK 和 Akt 的组合效应,并提供了一个框架来从机制上解释实验结果并指导实验设计。因此,该模型可用于研究针对 FGF 和 VEGF 刺激时 ERK 和 Akt 激活的促血管生成和抗血管生成治疗的影响。视频摘要。