Mayordomo-Cava Jennifer, Iborra-Lázaro Guillermo, Djebari Souhail, Temprano-Carazo Sara, Sánchez-Rodríguez Irene, Jeremic Danko, Gruart Agnès, Delgado-García José María, Jiménez-Díaz Lydia, Navarro-López Juan D
Neurophysiology and Behavioral Lab, Centro Regional de Investigaciones Biomédicas, School of Medicine of Ciudad Real, University of Castilla-La Mancha, 13071 Ciudad Real, Spain.
Division of Neurosciences, Pablo de Olavide University, 41013 Seville, Spain.
Biology (Basel). 2020 Jul 20;9(7):175. doi: 10.3390/biology9070175.
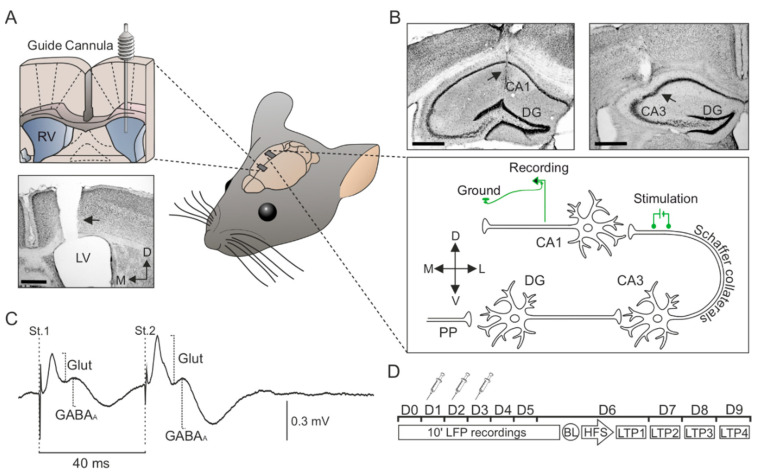
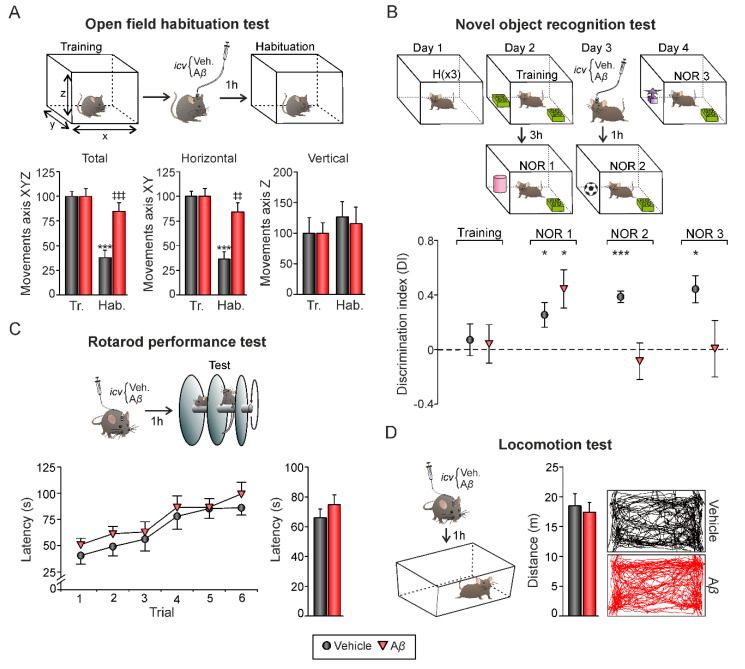
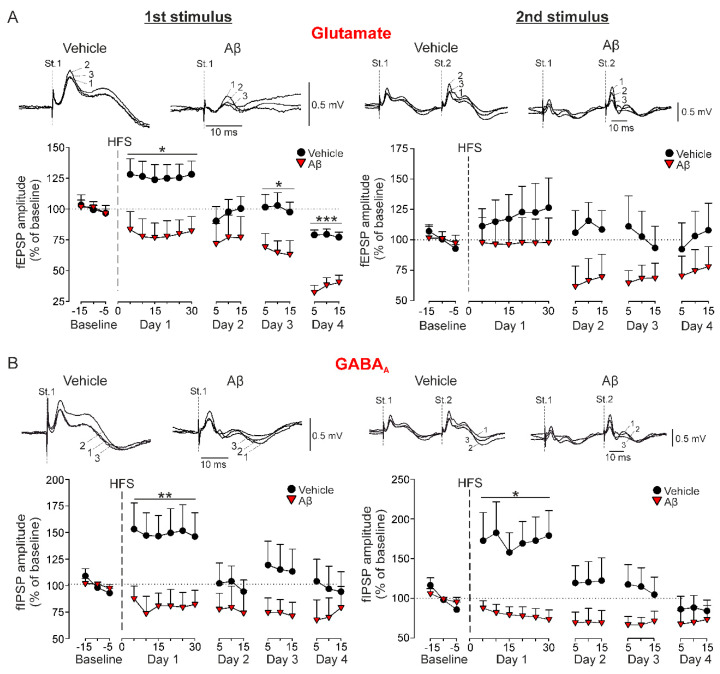
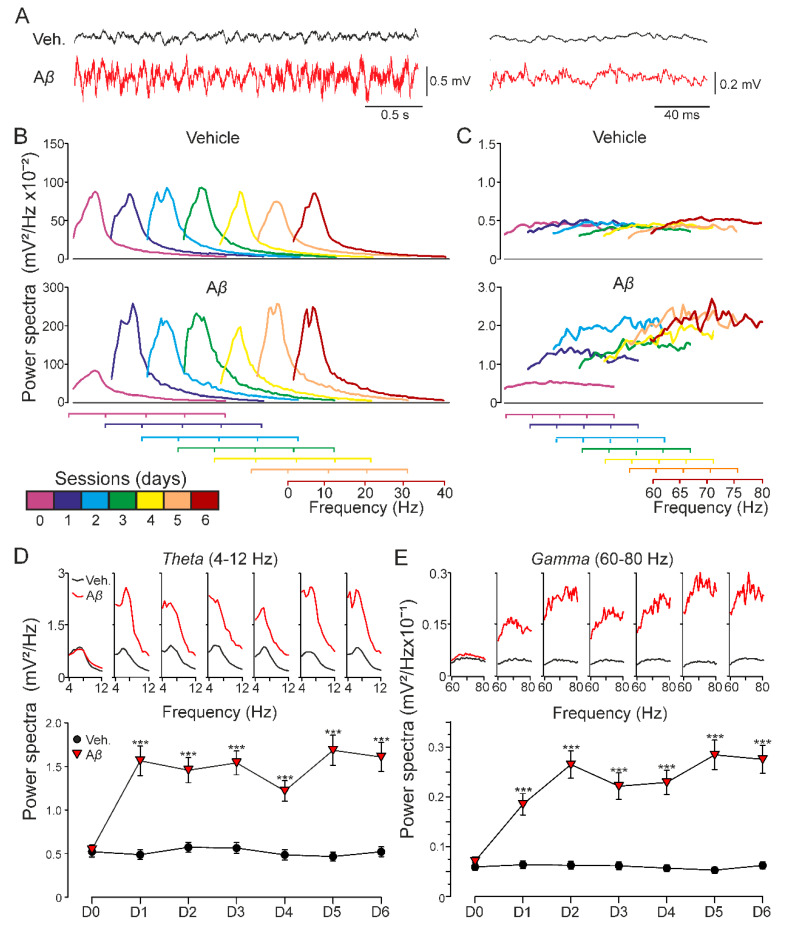
In early Alzheimer disease (AD) models synaptic failures and upstreaming aberrant patterns of network synchronous activity result in hippocampal-dependent memory deficits. In such initial stage, soluble forms of Amyloid- (A) peptides have been shown to play a causal role. Among different A species, A has been identified as the biologically active fragment, as induces major neuropathological signs related to early AD stages. Consequently, it has been extensively used to acutely explore the pathophysiological events related with neuronal dysfunction induced by soluble A forms. However, the synaptic mechanisms underlying its toxic effects on hippocampal-dependent memory remain unresolved. Here, in an in vivo model of amyloidosis generated by intracerebroventricular injections of A we studied the synaptic dysfunction mechanisms underlying hippocampal cognitive deficits. At the synaptic level, long-term potentiation (LTP) of synaptic excitation and inhibition was induced in CA1 region by high frequency simulation (HFS) applied to collaterals. A was found to alter metaplastic mechanisms of plasticity, facilitating long-term depression (LTD) of both types of LTP. In addition, aberrant synchronization of hippocampal network activity was found while at the behavioral level, deficits in hippocampal-dependent habituation and recognition memories emerged. Together, our results provide a substrate for synaptic disruption mechanism underlying hippocampal cognitive deficits present in A amyloidosis model.
在早期阿尔茨海默病(AD)模型中,突触功能障碍和网络同步活动的上游异常模式会导致海马体依赖性记忆缺陷。在这个初始阶段,可溶性淀粉样蛋白(A)肽已被证明起因果作用。在不同的A种类中,Aβ已被确定为生物活性片段,因为它会引发与早期AD阶段相关的主要神经病理学症状。因此,它已被广泛用于急性探索与可溶性A形式诱导的神经元功能障碍相关的病理生理事件。然而,其对海马体依赖性记忆产生毒性作用的突触机制仍未得到解决。在此,在通过脑室内注射Aβ产生的淀粉样变性体内模型中,我们研究了海马体认知缺陷背后的突触功能障碍机制。在突触水平上,通过对侧支施加高频刺激(HFS)在CA1区域诱导突触兴奋和抑制的长期增强(LTP)。发现Aβ会改变可塑性的元可塑性机制,促进两种类型LTP的长期抑制(LTD)。此外,发现海马体网络活动异常同步,而在行为水平上,出现了海马体依赖性习惯化和识别记忆缺陷。总之,我们的结果为Aβ淀粉样变性模型中存在的海马体认知缺陷背后的突触破坏机制提供了一个基础。