Mishra Jay S, Blesson Chellakkan S, Kumar Sathish
Department of Comparative Biosciences, School of Veterinary Medicine, University of Wisconsin, Madison, WI 53706, USA.
Reproductive Endocrinology and Infertility Division, Department of Obstetrics and Gynecology, Baylor College of Medicine and Family Fertility Center, Texas Children's Hospital, Houston, TX 77030, USA.
Biology (Basel). 2020 Jul 20;9(7):176. doi: 10.3390/biology9070176.
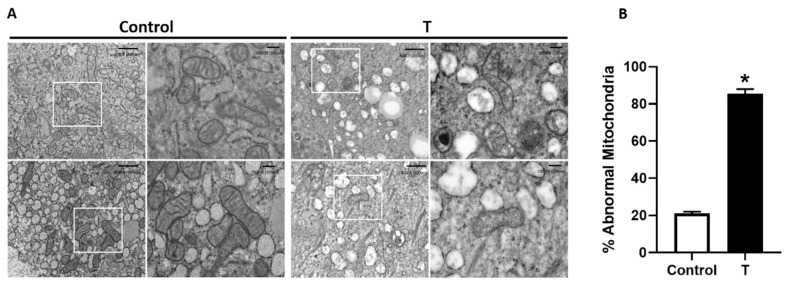
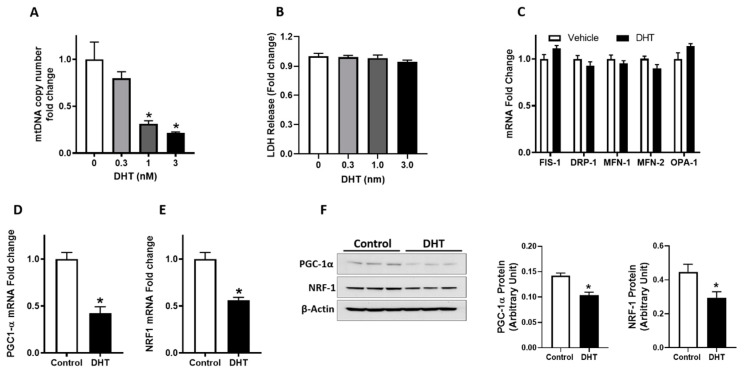
Placental mitochondrial dysfunction plays a central role in the pathogenesis of preeclampsia. Since preeclampsia is a hyperandrogenic state, we hypothesized that elevated maternal testosterone levels induce damage to placental mitochondria and decrease bioenergetic profiles. To test this hypothesis, pregnant Sprague-Dawley rats were injected with vehicle or testosterone propionate (0.5 mg/kg/day) from gestation day (GD) 15 to 19. On GD20, the placentas were isolated to assess mitochondrial structure, copy number, ATP/ADP ratio, and biogenesis (Pgc-1α and Nrf1). In addition, in vitro cultures of human trophoblasts (HTR-8/SVneo) were treated with dihydrotestosterone (0.3, 1.0, and 3.0 nM), and bioenergetic profiles using seahorse analyzer were assessed. Testosterone exposure in pregnant rats led to a 2-fold increase in plasma testosterone levels with an associated decrease in placental and fetal weights compared with controls. Elevated maternal testosterone levels induced structural damage to the placental mitochondria and decreased mitochondrial copy number. The ATP/ADP ratio was reduced with a parallel decrease in the mRNA and protein expression of Pgc-1α and Nrf1 in the placenta of testosterone-treated rats compared with controls. In cultured trophoblasts, dihydrotestosterone decreased the mitochondrial copy number and reduced PGC-1α, NRF1 mRNA, and protein levels without altering the expression of mitochondrial fission/fusion genes. Dihydrotestosterone exposure induced significant mitochondrial energy deficits with a dose-dependent decrease in basal respiration, ATP-linked respiration, maximal respiration, and spare respiratory capacity. In summary, our study suggests that the placental mitochondrial dysfunction induced by elevated maternal testosterone might be a potential mechanism linking preeclampsia to feto-placental growth restriction.
胎盘线粒体功能障碍在子痫前期的发病机制中起核心作用。由于子痫前期是一种高雄激素状态,我们推测母体睾酮水平升高会导致胎盘线粒体损伤并降低生物能量特征。为了验证这一假设,从妊娠第15天至19天,给怀孕的Sprague-Dawley大鼠注射溶剂或丙酸睾酮(0.5毫克/千克/天)。在妊娠第20天,分离胎盘以评估线粒体结构、拷贝数、ATP/ADP比值和生物发生(Pgc-1α和Nrf1)。此外,用人滋养层细胞(HTR-8/SVneo)进行体外培养,用二氢睾酮(0.3、1.0和3.0纳摩尔)处理,并使用海马分析仪评估生物能量特征。与对照组相比,怀孕大鼠暴露于睾酮导致血浆睾酮水平增加2倍,同时胎盘和胎儿重量下降。母体睾酮水平升高导致胎盘线粒体结构损伤并降低线粒体拷贝数。与对照组相比,睾酮处理大鼠胎盘的ATP/ADP比值降低,同时Pgc-1α和Nrf1的mRNA和蛋白表达平行下降。在培养的滋养层细胞中,二氢睾酮降低线粒体拷贝数并降低PGC-1α、NRF1 mRNA和蛋白水平,而不改变线粒体分裂/融合基因的表达。二氢睾酮暴露导致明显的线粒体能量缺陷,基础呼吸、ATP相关呼吸、最大呼吸和备用呼吸能力呈剂量依赖性下降。总之,我们的研究表明,母体睾酮升高诱导的胎盘线粒体功能障碍可能是将子痫前期与胎儿-胎盘生长受限联系起来的潜在机制。