The Jackson Laboratory, 600 Main Street, Bar Harbor, 04609, ME, USA.
University of Michigan, 500 South State Street, Ann Arbor, 48109, MI, USA.
Genome Biol. 2020 Jul 27;21(1):183. doi: 10.1186/s13059-020-02103-2.
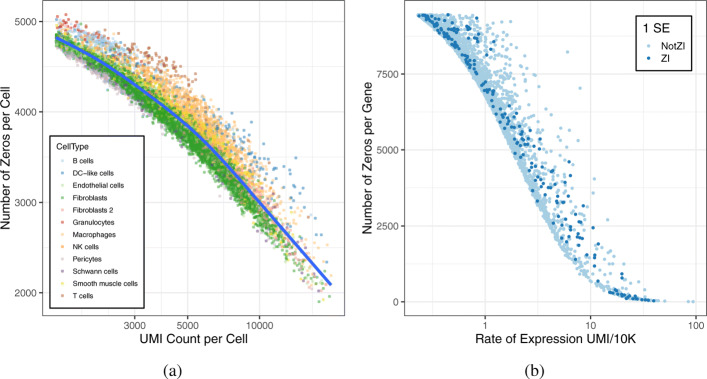
Single-cell RNA sequencing is a powerful tool for characterizing cellular heterogeneity in gene expression. However, high variability and a large number of zero counts present challenges for analysis and interpretation. There is substantial controversy over the origins and proper treatment of zeros and no consensus on whether zero-inflated count distributions are necessary or even useful. While some studies assume the existence of zero inflation due to technical artifacts and attempt to impute the missing information, other recent studies argue that there is no zero inflation in scRNA-seq data.
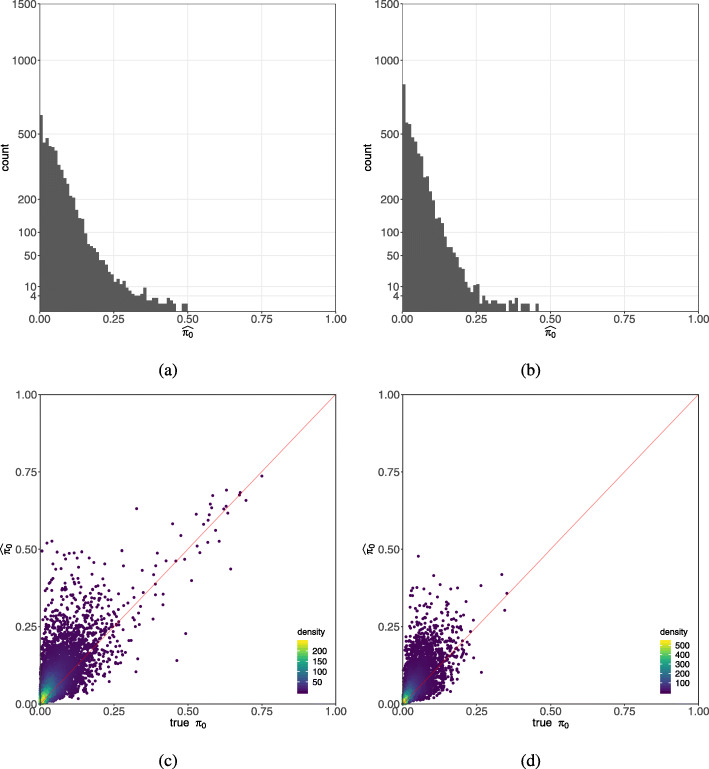
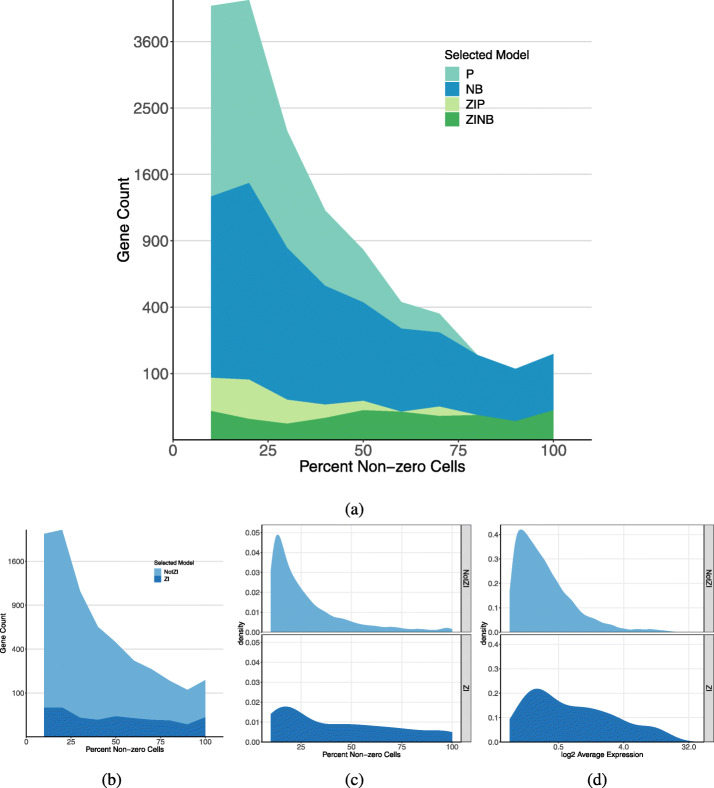
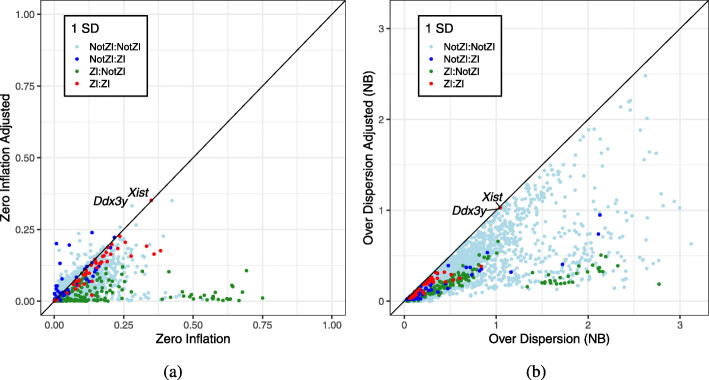
We apply a Bayesian model selection approach to unambiguously demonstrate zero inflation in multiple biologically realistic scRNA-seq datasets. We show that the primary causes of zero inflation are not technical but rather biological in nature. We also demonstrate that parameter estimates from the zero-inflated negative binomial distribution are an unreliable indicator of zero inflation.
Despite the existence of zero inflation in scRNA-seq counts, we recommend the generalized linear model with negative binomial count distribution, not zero-inflated, as a suitable reference model for scRNA-seq analysis.
单细胞 RNA 测序是一种强大的工具,可用于描述基因表达中的细胞异质性。然而,高变异性和大量的零计数给分析和解释带来了挑战。关于零的起源和正确处理存在很大争议,对于是否需要零膨胀计数分布,甚至是否有用,也没有共识。虽然有些研究由于技术因素假设存在零膨胀,并尝试推断缺失信息,但其他最近的研究则认为 scRNA-seq 数据中没有零膨胀。
我们应用贝叶斯模型选择方法明确证明了多个生物学上合理的 scRNA-seq 数据集存在零膨胀。我们表明,零膨胀的主要原因不是技术上的,而是生物学上的。我们还表明,零膨胀负二项分布的参数估计是零膨胀的不可靠指标。
尽管 scRNA-seq 计数中存在零膨胀,但我们建议使用具有负二项计数分布的广义线性模型,而不是零膨胀,作为 scRNA-seq 分析的合适参考模型。