Mehta Supriya D, Nannini Drew R, Otieno Fredrick, Green Stefan J, Agingu Walter, Landay Alan, Zheng Yinan, Hou Lifang
Division of Epidemiology & Biostatistics, University of Illinois at Chicago School of Public Health, Chicago, Illinois, USA
Center for Global Oncology, Institute of Global Health, Northwestern University Feinberg School of Medicine, Chicago, Illinois, USA.
mSystems. 2020 Jul 28;5(4):e00502-20. doi: 10.1128/mSystems.00502-20.
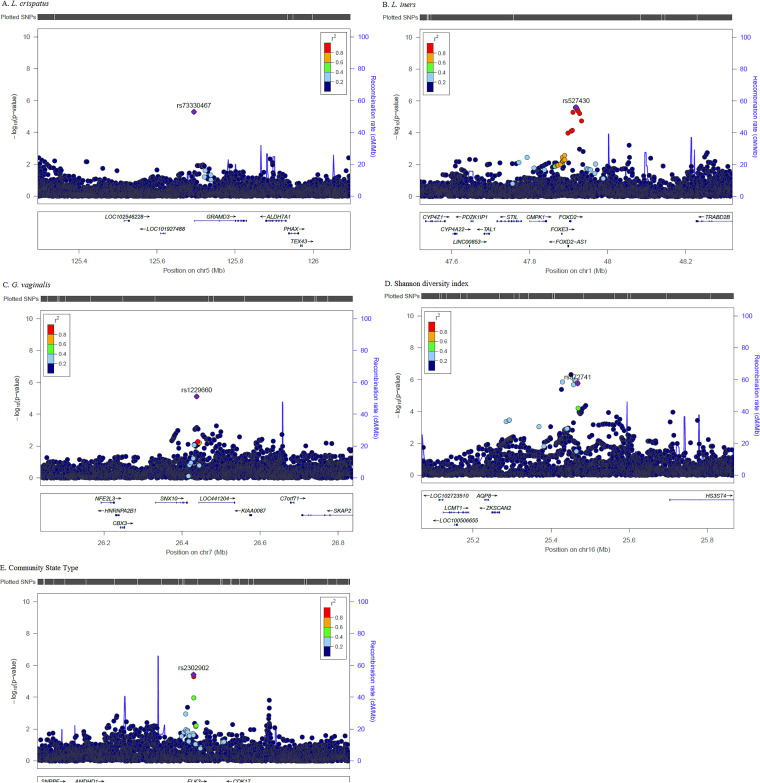
Bacterial vaginosis (BV) affects 20% of women worldwide and is associated with adverse reproductive health outcomes and increased risk for HIV. Typically, BV represents a shift in the vaginal microbiome from one that is dominated by to one that is diverse. Persistent racial differences in BV and diverse vaginal microbiome composition overlap with racial disparities in risks for HIV and sexually transmitted infection, especially among women of African descent. Risk factors for BV and nonoptimal vaginal microbiome include sexual practices, yet racial differences persist when adjusted for behavioral factors, suggesting a host genetic component. Here, we perform a genome-wide association study on vaginal microbiome traits in Kenyan women. Linear regression and logistic regression were performed, adjusting for age and principal components of genetic ancestry, to evaluate the association between , , , Shannon diversity index, and community state type (CST) with host genetic single nucleotide polymorphisms (SNPs). We identified novel genomic loci associated with the vaginal microbiome traits, though no SNP reached genome-wide significance. During pathway enrichment analysis, Toll-like receptors (TLRs), cytokine production, and other components of innate immune response were associated with , , and CST. Multiple previously reported genomic loci were replicated, including (Shannon, CST), (, Shannon), (Shannon, CST), (, , CST), and (, Shannon). These genetic associations suggest a role for the innate immune system and cell signaling in vaginal microbiome composition and susceptibility to nonoptimal vaginal microbiome. Globally, bacterial vaginosis (BV) is a common condition in women. BV is associated with poorer reproductive health outcomes and HIV risk. Typically, BV represents a shift in the vaginal microbiome from one that is dominated by to one that is diverse. Despite many women having similar exposures, the prevalence of BV and nonoptimal vaginal microbiome is increased for women of African descent, suggesting a possible role for host genetics. We conducted a genome-wide association study of important vaginal microbiome traits in Kenyan women. We identified novel genetic loci and biological pathways related to mucosal immunity, cell signaling, and infection that were associated with vaginal microbiome traits; we replicated previously reported loci associated with mucosal immune response. These results provide insight into potential host genetic influences on vaginal microbiome composition and can guide larger longitudinal studies, with genetic and functional comparison across microbiome sites within individuals and across populations.
细菌性阴道病(BV)影响着全球20%的女性,与不良生殖健康结局及感染HIV的风险增加相关。通常,BV表现为阴道微生物群从以某一种微生物为主导转变为多种微生物共存。BV及多样的阴道微生物群组成方面持续存在的种族差异,与HIV及性传播感染风险方面的种族差异相重叠,尤其是在非洲裔女性中。BV及非最佳阴道微生物群的风险因素包括性行为,但在对行为因素进行校正后种族差异依然存在,这表明存在宿主遗传因素。在此,我们对肯尼亚女性的阴道微生物群特征进行了全基因组关联研究。进行了线性回归和逻辑回归分析,并对年龄和遗传血统的主成分进行了校正,以评估某一微生物、某一种微生物丰度、某一种微生物相对丰度、香农多样性指数以及群落状态类型(CST)与宿主遗传单核苷酸多态性(SNP)之间的关联。我们鉴定出了与阴道微生物群特征相关的新基因组位点,尽管没有SNP达到全基因组显著性水平。在通路富集分析过程中,Toll样受体(TLRs)、细胞因子产生以及先天性免疫反应的其他成分与某一微生物、某一种微生物丰度和CST相关。多个先前报道的基因组位点得到了重复验证,包括某一基因(香农指数、CST)、某一基因(某一种微生物、香农指数)、某一基因(香农指数、CST)、某一基因(某一种微生物、某一种微生物丰度、CST)以及某一基因(某一种微生物、香农指数)。这些遗传关联表明先天性免疫系统和细胞信号传导在阴道微生物群组成以及对非最佳阴道微生物群的易感性中发挥作用。在全球范围内,细菌性阴道病(BV)是女性中的一种常见病症。BV与较差的生殖健康结局及HIV感染风险相关。通常,BV表现为阴道微生物群从以某一种微生物为主导转变为多种微生物共存。尽管许多女性有相似的暴露情况,但非洲裔女性中BV及非最佳阴道微生物群的患病率更高,这表明宿主遗传学可能发挥作用。我们对肯尼亚女性重要的阴道微生物群特征进行了全基因组关联研究。我们鉴定出了与阴道微生物群特征相关的新基因位点以及与黏膜免疫、细胞信号传导和感染相关的生物学通路;我们重复验证了先前报道的与黏膜免疫反应相关的位点。这些结果为宿主遗传因素对阴道微生物群组成的潜在影响提供了见解,并可指导更大规模的纵向研究,在个体内部以及不同人群之间进行微生物群位点的遗传和功能比较。