Huntsman Cancer Institute, University of Utah, Salt Lake City, UT 84112, USA.
Huntsman Cancer Institute, University of Utah, Salt Lake City, UT 84112, USA; Department of Oncological Sciences, University of Utah, Salt Lake City, UT 84112, USA.
Cell Rep. 2020 Aug 4;32(5):107994. doi: 10.1016/j.celrep.2020.107994.
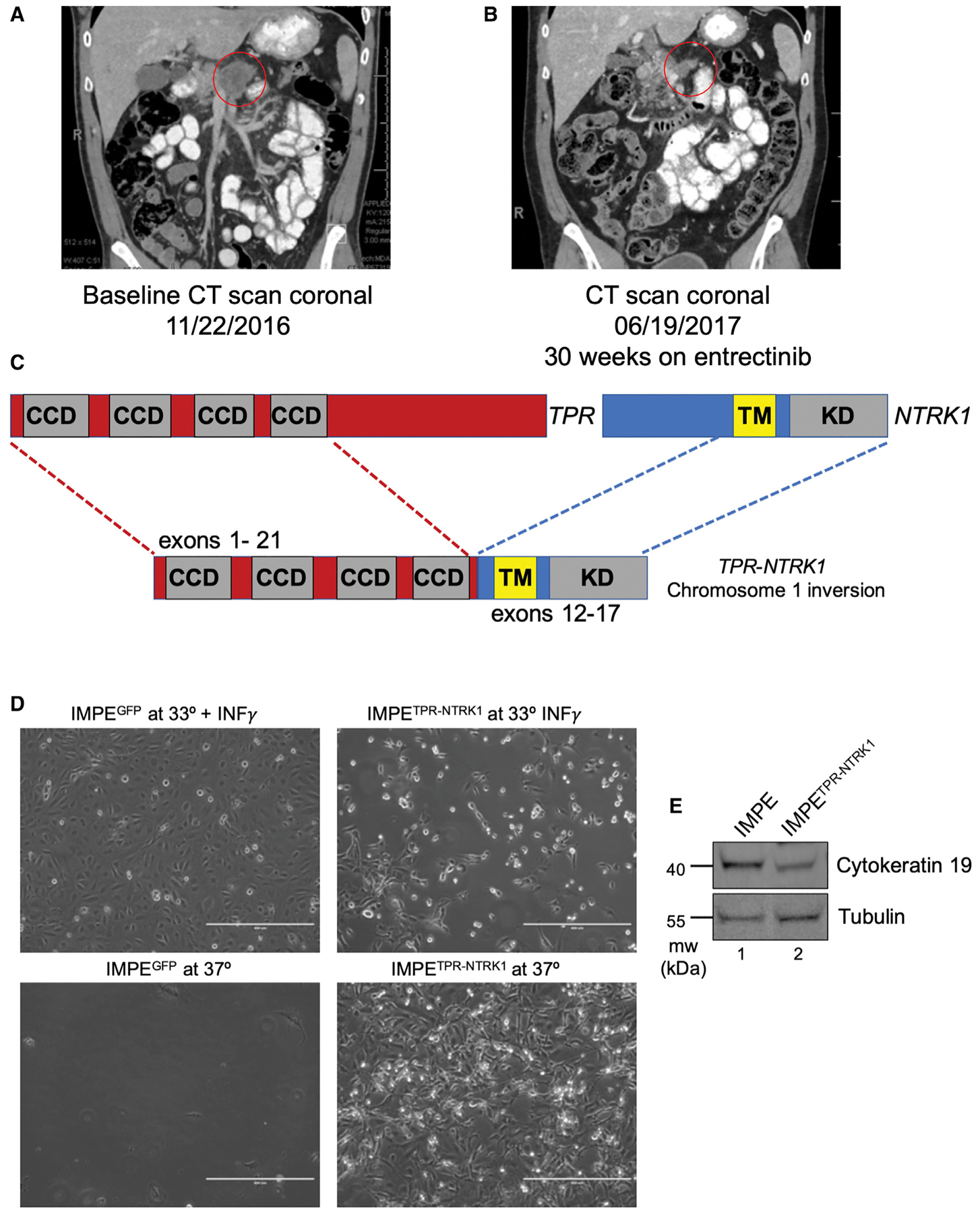
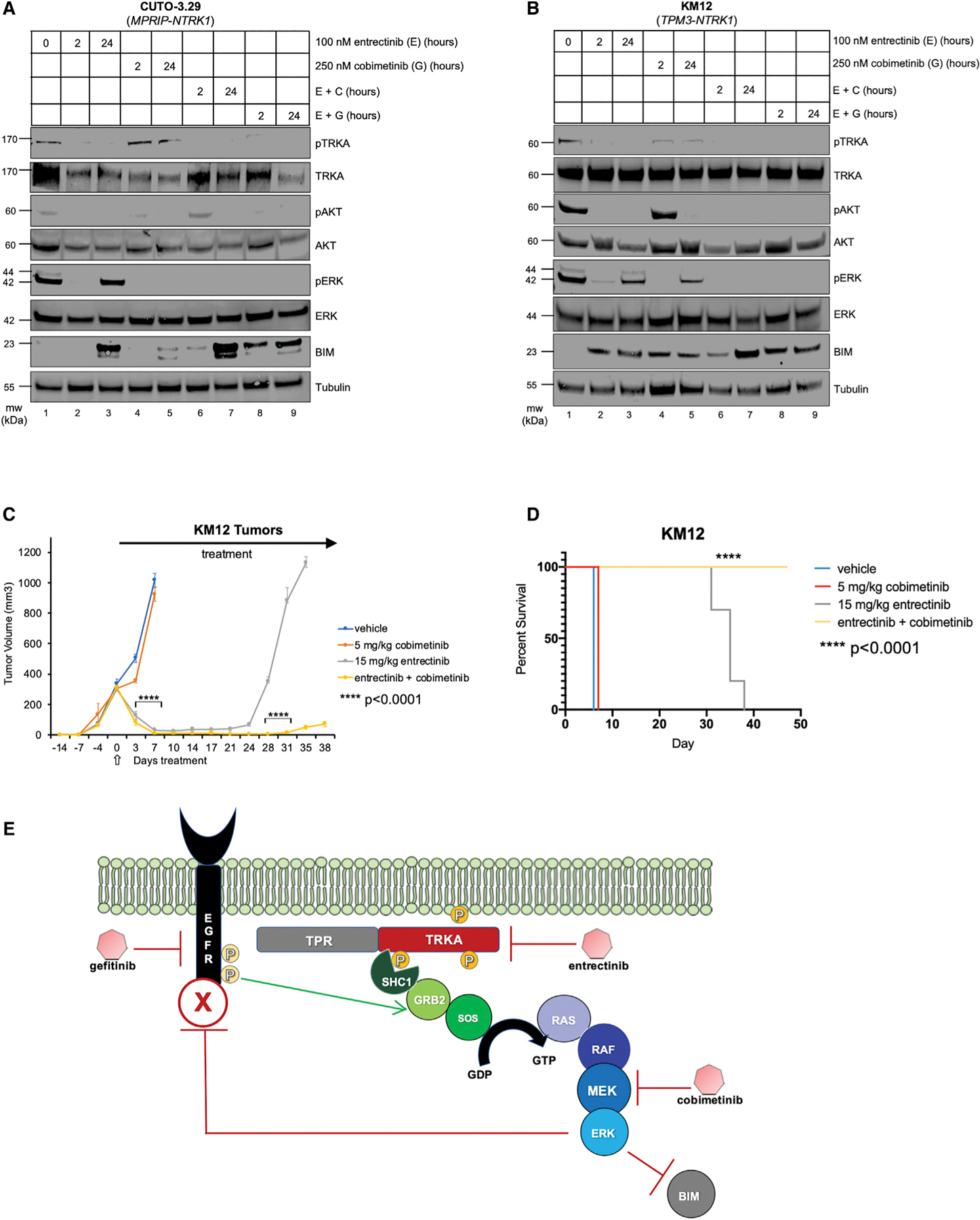
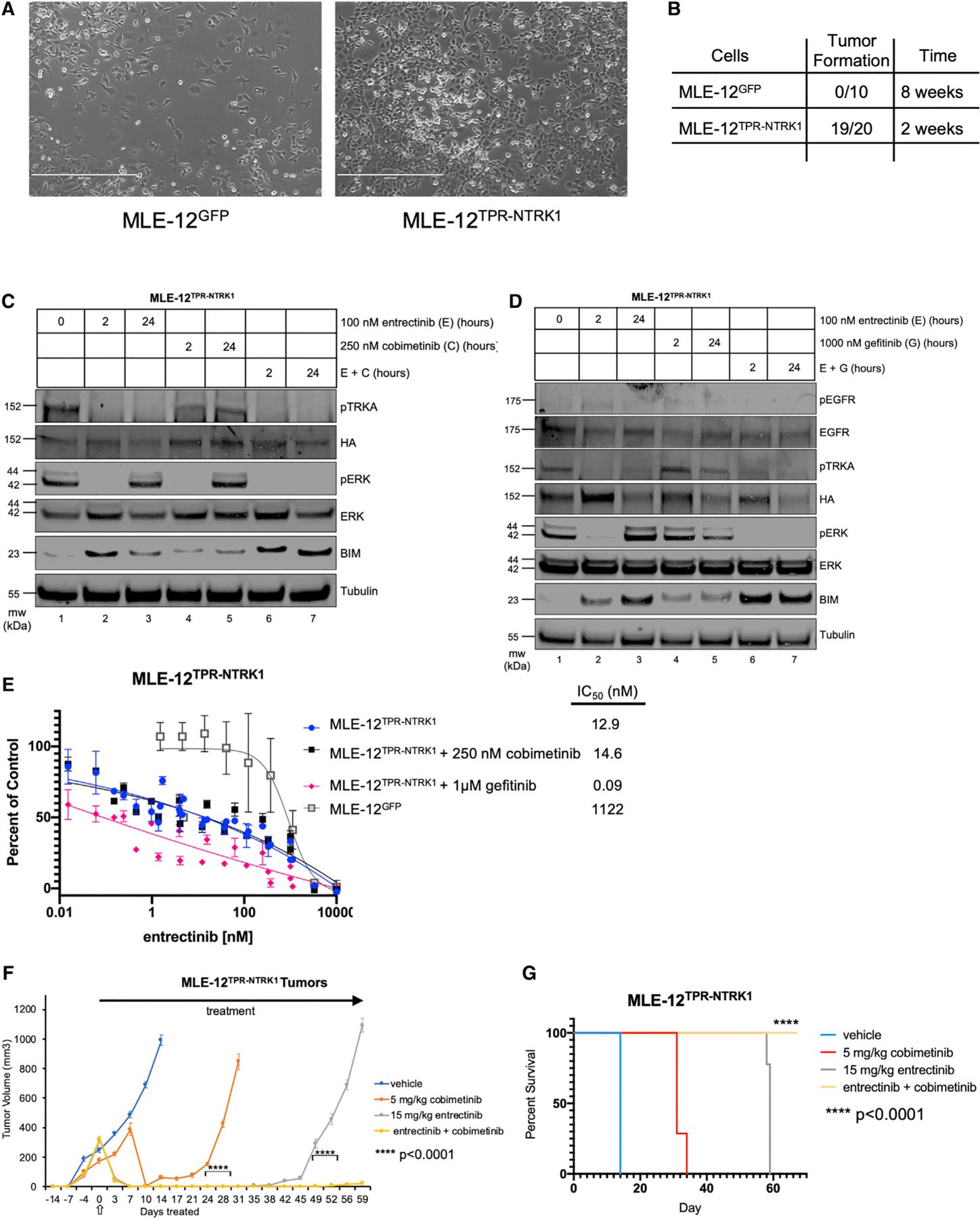
NTRK1 gene fusions are actionable drivers of numerous human malignancies. Here, we show that expression of the TPR-NTRK1 fusion kinase in immortalized mouse pancreatic ductal epithelial (IMPE) (pancreas) or mouse lung epithelial (MLE-12) cells is sufficient to promote rapidly growing tumors in mice. Both tumor models are exquisitely sensitive to targeted inhibition with entrectinib, a tropomyosin-related kinase A (TRKA) inhibitor. Initial regression of NTRK1-driven tumors is driven by induced expression of BIM, such that BIM silencing leads to a diminished response to entrectinib in vivo. However, the emergence of drug-resistant disease limits the long-term durability of responses. Based on the reactivation of RAF>MEK>ERK signaling observed in entrectinib-treated tumors, we show that the combination of entrectinib plus the MEK1/2 inhibitor cobimetinib dramatically forestalls the onset of drug resistance in vivo. Collectively, these data provide a mechanistic rationale for rapid clinical deployment of combined inhibition of TRKA plus MEK1/2 in NTRK1-driven cancers.
NTRK1 基因融合是许多人类恶性肿瘤的可操作驱动因素。在这里,我们表明,TPR-NTRK1 融合激酶在永生化的小鼠胰腺导管上皮(IMPE)(胰腺)或小鼠肺上皮(MLE-12)细胞中的表达足以促进小鼠中快速生长的肿瘤。这两种肿瘤模型对靶向抑制剂 entrectinib(一种原肌球蛋白相关激酶 A(TRKA)抑制剂)非常敏感。在 NTRK1 驱动的肿瘤中,最初的肿瘤消退是由 BIM 的诱导表达驱动的,因此 BIM 沉默会导致体内对 entrectinib 的反应减弱。然而,耐药性疾病的出现限制了反应的长期持久性。基于在 entrectinib 治疗的肿瘤中观察到的 RAF>MEK>ERK 信号的重新激活,我们表明,entrectinib 加 MEK1/2 抑制剂 cobimetinib 的联合使用可在体内显著阻止耐药性的发生。总的来说,这些数据为在 NTRK1 驱动的癌症中快速临床应用联合抑制 TRKA 加 MEK1/2 提供了机制基础。