Yasuda Makiko, Huston Marshall W, Pagant Silvere, Gan Lin, St Martin Susan, Sproul Scott, Richards Daniel, Ballaron Stephen, Hettini Khaled, Ledeboer Annemarie, Falese Lillian, Cao Liching, Lu Yanmei, Holmes Michael C, Meyer Kathleen, Desnick Robert J, Wechsler Thomas
Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, USA.
Sangamo Therapeutics, Inc., Brisbane, CA 94005, USA.
Mol Ther Methods Clin Dev. 2020 Jul 9;18:607-619. doi: 10.1016/j.omtm.2020.07.002. eCollection 2020 Sep 11.
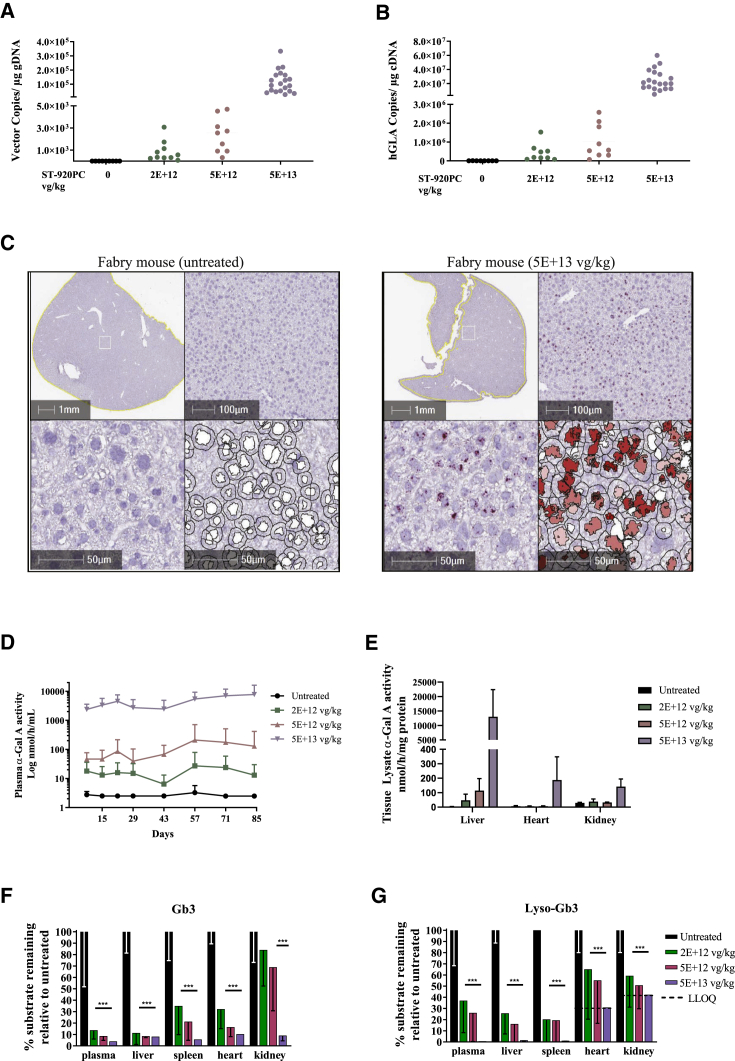
Fabry disease is an X-linked lysosomal storage disorder caused by mutations in the alpha-galactosidase A () gene, which encodes the exogalactosyl hydrolase, alpha-galactosidase A (α-Gal A). Deficient α-Gal A activity results in the progressive, systemic accumulation of its substrates, globotriaosylceramide (Gb3) and globotriaosylsphingosine (Lyso-Gb3), leading to renal, cardiac, and/or cerebrovascular disease and early demise. The current standard treatment for Fabry disease is enzyme replacement therapy, which necessitates lifelong biweekly infusions of recombinant enzyme. A more long-lasting treatment would benefit Fabry patients. Here, a gene therapy approach using an episomal adeno-associated viral 2/6 (AAV2/6) vector that encodes the human cDNA driven by a liver-specific expression cassette was evaluated in a Fabry mouse model that lacks α-Gal A activity and progressively accumulates Gb3 and Lyso-Gb3 in plasma and tissues. A detailed 3-month pharmacology and toxicology study showed that administration of a clinical-scale-manufactured AAV2/6 vector resulted in markedly increased plasma and tissue α-Gal A activities, and essentially normalized Gb3 and Lyso-Gb3 at key sites of pathology. Further optimization of vector design identified the clinical lead vector, ST-920, which produced several-fold higher plasma and tissue α-Gal A activity levels with a good safety profile. Together, these studies provide the basis for the clinical development of ST-920.
法布里病是一种X连锁溶酶体贮积症,由α-半乳糖苷酶A()基因突变引起,该基因编码外切半乳糖基水解酶α-半乳糖苷酶A(α-Gal A)。α-Gal A活性不足导致其底物球三糖神经酰胺(Gb3)和球三糖鞘氨醇(Lyso-Gb3)在全身进行性蓄积,从而导致肾脏、心脏和/或脑血管疾病以及过早死亡。法布里病目前的标准治疗方法是酶替代疗法,需要每两周进行一次终身重组酶输注。一种更持久的治疗方法将使法布里病患者受益。在此,在缺乏α-Gal A活性且血浆和组织中Gb3和Lyso-Gb3逐渐蓄积的法布里病小鼠模型中,评估了一种使用游离型腺相关病毒2/6(AAV2/6)载体的基因治疗方法,该载体由肝脏特异性表达盒驱动,编码人 cDNA。一项详细的为期3个月的药理学和毒理学研究表明,给予临床规模生产的AAV2/6载体可使血浆和组织中的α-Gal A活性显著增加,并使病理关键部位的Gb3和Lyso-Gb3基本恢复正常。载体设计的进一步优化确定了临床主导载体ST-920,其血浆和组织α-Gal A活性水平提高了数倍,且安全性良好。总之,这些研究为ST-920的临床开发提供了依据。