Etich Julia, Leßmeier Lennart, Rehberg Mirko, Sill Helge, Zaucke Frank, Netzer Christian, Semler Oliver
Dr. Rolf M. Schwiete Research Unit for Osteoarthritis, Orthopedic University Hospital Friedrichsheim gGmbH, Frankfurt/Main, Germany.
University of Cologne, Faculty of Medicine and University Hospital Cologne, Institute of Human Genetics, Cologne, Germany.
Mol Cell Pediatr. 2020 Aug 14;7(1):9. doi: 10.1186/s40348-020-00101-9.
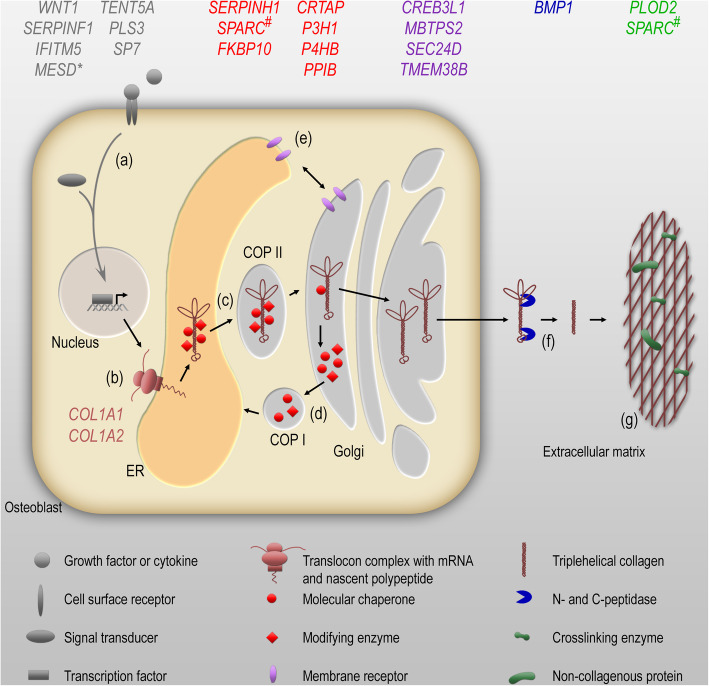
Osteogenesis imperfecta (OI) is a rare congenital disease with a wide spectrum of severity characterized by skeletal deformity and increased bone fragility as well as additional, variable extraskeletal symptoms. Here, we present an overview of the genetic heterogeneity and pathophysiological background of OI as well as OI-related bone fragility disorders and highlight current therapeutic options.The most common form of OI is caused by mutations in the two collagen type I genes. Stop mutations usually lead to reduced collagen amount resulting in a mild phenotype, while missense mutations mainly provoke structural alterations in the collagen protein and entail a more severe phenotype. Numerous other causal genes have been identified during the last decade that are involved in collagen biosynthesis, modification and secretion, the differentiation and function of osteoblasts, and the maintenance of bone homeostasis.Management of patients with OI involves medical treatment by bisphosphonates as the most promising therapy to inhibit bone resorption and thereby facilitate bone formation. Surgical treatment ensures pain reduction and healing without an increase of deformities. Timely remobilization and regular strengthening of the muscles by physiotherapy are crucial to improve mobility, prevent muscle wasting and avoid bone resorption caused by immobilization. Identification of the pathomechanism for SERPINF1 mutations led to the development of a tailored mechanism-based therapy using denosumab, and unraveling further pathomechanisms will likely open new avenues for innovative treatment approaches.
成骨不全症(OI)是一种罕见的先天性疾病,严重程度范围广泛,其特征为骨骼畸形、骨脆性增加以及其他各种不同的骨骼外症状。在此,我们概述了成骨不全症的遗传异质性和病理生理背景以及与成骨不全症相关的骨脆性疾病,并着重介绍了当前的治疗选择。成骨不全症最常见的形式是由两个I型胶原蛋白基因突变引起的。终止突变通常导致胶原蛋白数量减少,从而产生轻度表型,而错义突变主要引发胶原蛋白蛋白质的结构改变,并导致更严重的表型。在过去十年中,已鉴定出许多其他致病基因,它们参与胶原蛋白的生物合成、修饰和分泌、成骨细胞的分化和功能以及骨稳态的维持。成骨不全症患者的管理包括使用双膦酸盐进行药物治疗,这是抑制骨吸收从而促进骨形成的最有前景的疗法。手术治疗可确保减轻疼痛并促进愈合,同时不会增加畸形。通过物理治疗及时进行活动恢复和定期增强肌肉力量对于改善活动能力、预防肌肉萎缩以及避免因固定不动导致的骨吸收至关重要。对丝氨酸蛋白酶抑制剂F1(SERPINF1)突变致病机制的识别促使开发了一种使用地诺单抗的基于机制的定制疗法,进一步揭示致病机制可能会为创新治疗方法开辟新途径。