Department of Statistics, University of Washington, Seattle, Washington, United States of America.
Computational Biology, Fred Hutchinson Cancer Research Center, Seattle, Washington, United States of America.
PLoS Comput Biol. 2020 Aug 17;16(8):e1008030. doi: 10.1371/journal.pcbi.1008030. eCollection 2020 Aug.
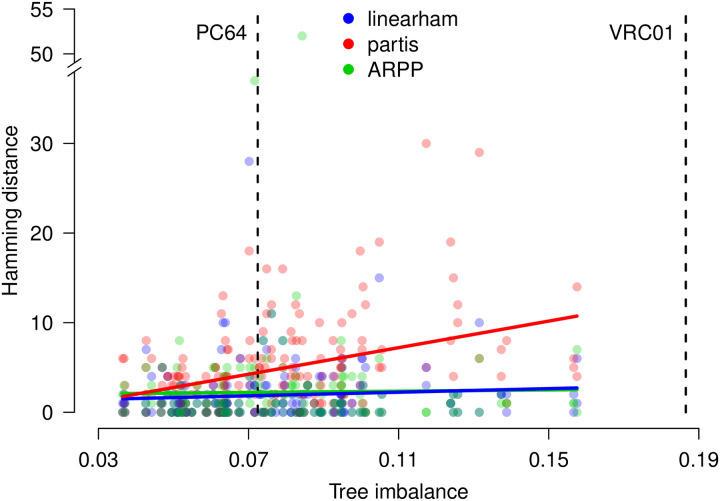
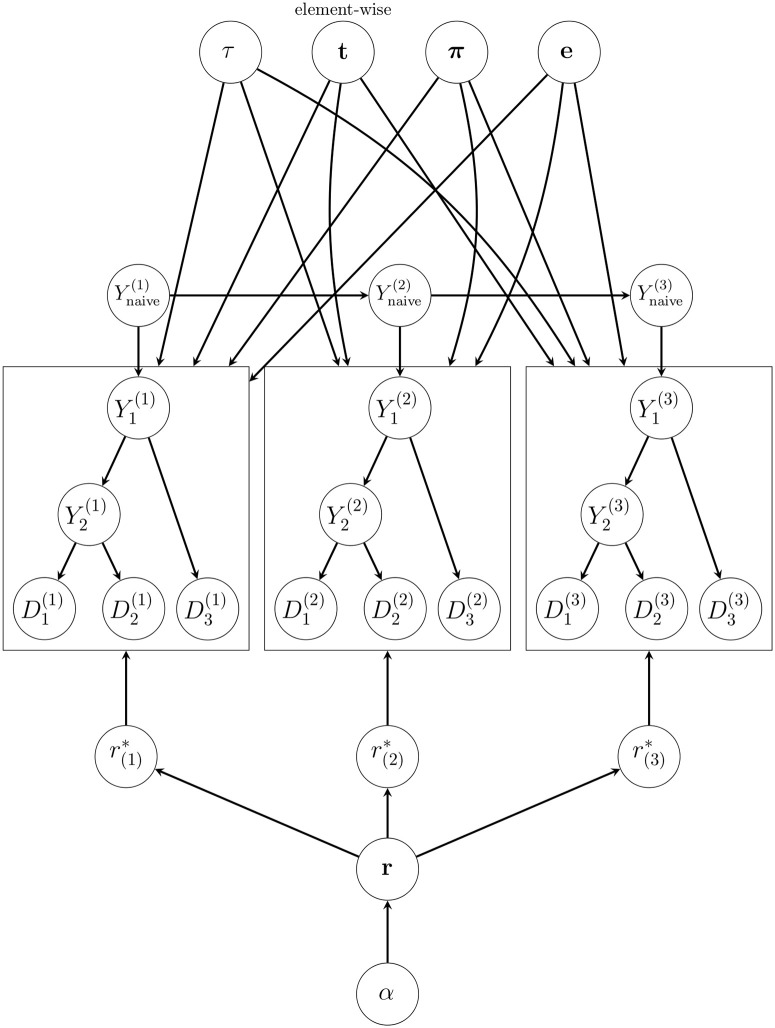
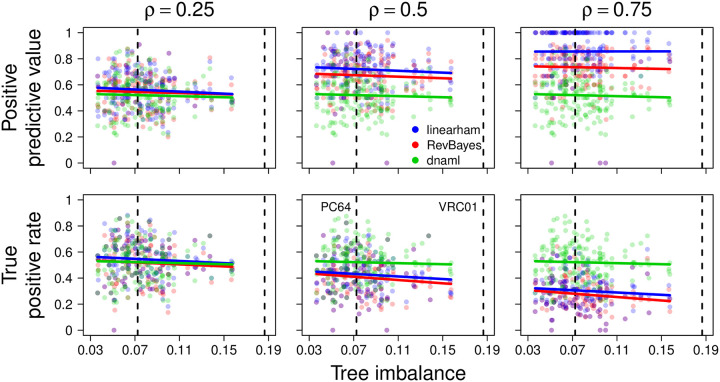
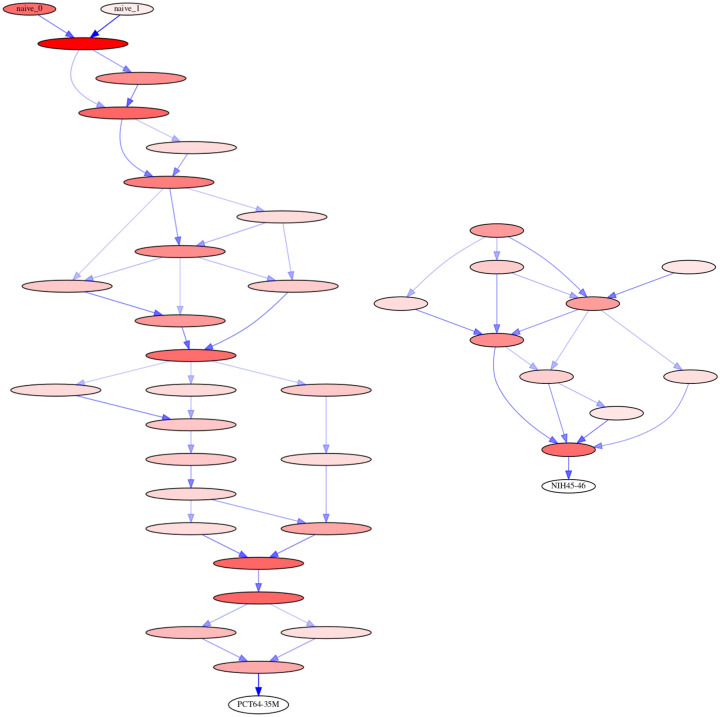
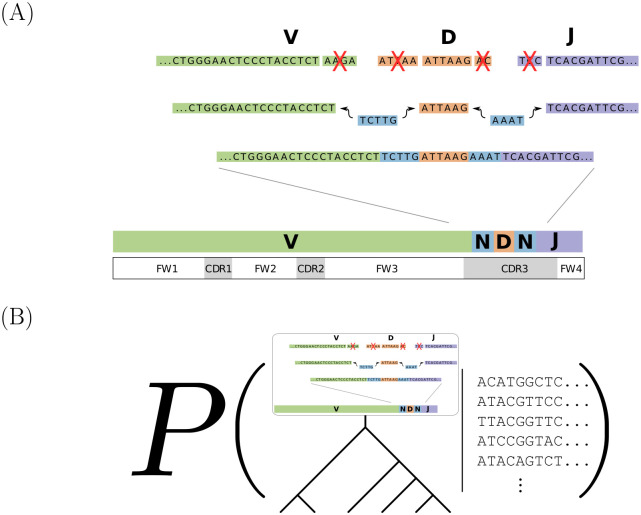
The human body generates a diverse set of high affinity antibodies, the soluble form of B cell receptors (BCRs), that bind to and neutralize invading pathogens. The natural development of BCRs must be understood in order to design vaccines for highly mutable pathogens such as influenza and HIV. BCR diversity is induced by naturally occurring combinatorial "V(D)J" rearrangement, mutation, and selection processes. Most current methods for BCR sequence analysis focus on separately modeling the above processes. Statistical phylogenetic methods are often used to model the mutational dynamics of BCR sequence data, but these techniques do not consider all the complexities associated with B cell diversification such as the V(D)J rearrangement process. In particular, standard phylogenetic approaches assume the DNA bases of the progenitor (or "naive") sequence arise independently and according to the same distribution, ignoring the complexities of V(D)J rearrangement. In this paper, we introduce a novel approach to Bayesian phylogenetic inference for BCR sequences that is based on a phylogenetic hidden Markov model (phylo-HMM). This technique not only integrates a naive rearrangement model with a phylogenetic model for BCR sequence evolution but also naturally accounts for uncertainty in all unobserved variables, including the phylogenetic tree, via posterior distribution sampling.
人体产生了多种高亲和力的抗体,即 B 细胞受体 (BCR) 的可溶性形式,这些抗体可以结合并中和入侵的病原体。为了设计针对流感和艾滋病毒等高度易变病原体的疫苗,必须了解 BCR 的自然发育过程。BCR 的多样性是由自然发生的组合“V(D)J”重排、突变和选择过程诱导的。目前大多数 BCR 序列分析方法都侧重于分别对上述过程进行建模。统计系统发育方法通常用于对 BCR 序列数据的突变动态进行建模,但这些技术并未考虑与 B 细胞多样化相关的所有复杂性,例如 V(D)J 重排过程。特别是,标准系统发育方法假设祖代(或“原始”)序列的 DNA 碱基是独立产生的,并且符合相同的分布,而忽略了 V(D)J 重排的复杂性。在本文中,我们引入了一种基于系统发育隐马尔可夫模型 (phylogeny-HMM) 的 BCR 序列贝叶斯系统发育推断的新方法。该技术不仅将原始重排模型与 BCR 序列进化的系统发育模型相结合,而且还通过后验分布采样自然地考虑了所有未观察变量的不确定性,包括系统发育树。