Center for Translational Medicine, Department of Medicine, Sidney Kimmel Medical College, Thomas Jefferson University, Philadelphia, PA, 19107, USA.
Univ Lyon, CarMeN Laboratory, INSERM, INRA, INSA Lyon, Université Claude Bernard Lyon 1, 69500, Bron, France.
Cell Death Dis. 2020 Aug 19;11(8):661. doi: 10.1038/s41419-020-02864-5.
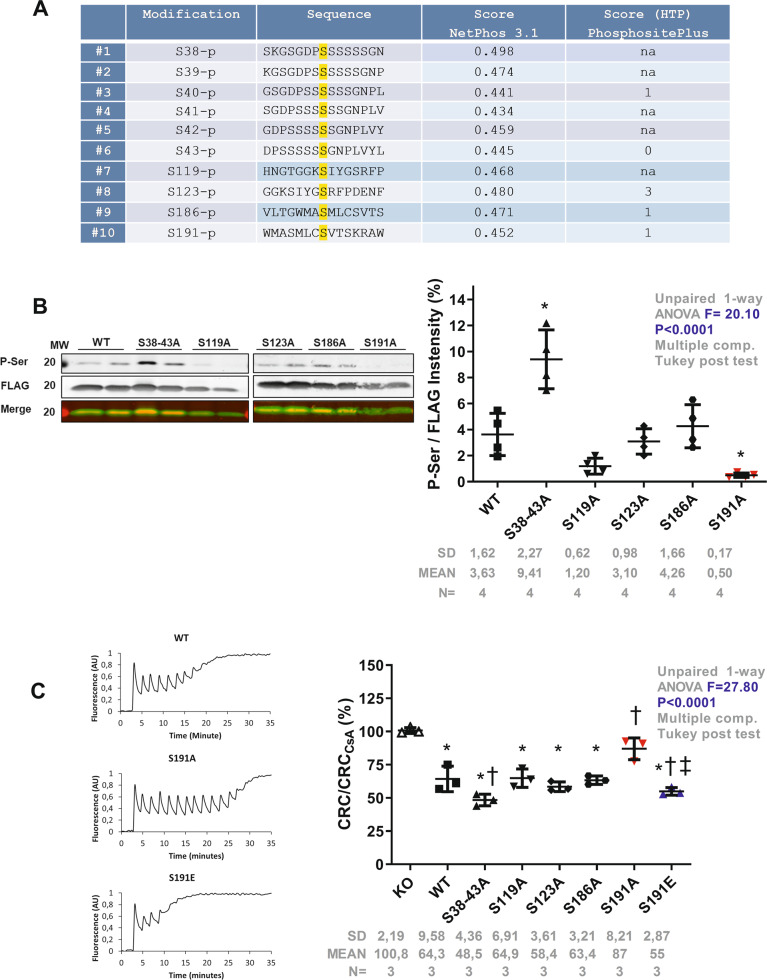
The mitochondrial permeability transition pore (mPTP) plays a critical role in the pathogenesis of cardiovascular diseases, including ischemia/reperfusion injury. Although the pore structure is still unresolved, the mechanism through which cyclophilin D (CypD) regulates mPTP opening is the subject of intensive studies. While post-translational modifications of CypD have been shown to modulate pore opening, specific phosphorylation sites of CypD have not yet been identified. We hypothesized here that phosphorylation of CypD on a serine residue controls mPTP opening and subsequent cell death at reperfusion. We combined in silico analysis with in vitro and genetic manipulations to determine potential CypD phosphorylation sites and their effect on mitochondrial function and cell death. Importantly, we developed an in vivo intramyocardial adenoviral strategy to assess the effect of the CypD phosphorylation event on infarct size. Our results show that although CypD can potentially be phosphorylated at multiple serine residues, only the phosphorylation status at S191 directly impacts the ability of CypD to regulate the mPTP. Protein-protein interaction strategies showed that the interaction between CypD and oligomycin sensitivity-conferring protein (OSCP) was reduced by 45% in the phosphoresistant S191A mutant, whereas it was increased by 48% in the phosphomimetic S191E mutant cells. As a result, the phosphoresistant CypD S191A mutant was protected against 18 h starvation whereas cell death was significantly increased in phosphomimetic S191E group, associated with mitochondrial respiration alteration and ROS production. As in vivo proof of concept, in S191A phosphoresistant rescued CypD-KO mice developed significantly smaller infarct as compared to WT whereas infarct size was drastically increased in S191E phosphomimetic rescued mice. We conclude that CypD phosphorylation at S191 residue leads to its binding to OSCP and thus sensitizes mPTP opening for the subsequent cell death.
线粒体通透性转换孔 (mPTP) 在包括缺血/再灌注损伤在内的心血管疾病发病机制中发挥着关键作用。尽管孔结构仍未得到解决,但环孢素 D (CypD) 调节 mPTP 开放的机制是深入研究的主题。虽然已经表明 CypD 的翻译后修饰可以调节孔的开放,但 CypD 的特定磷酸化位点尚未确定。我们在这里假设 CypD 丝氨酸残基的磷酸化控制着再灌注时 mPTP 的开放和随后的细胞死亡。我们结合了计算机分析、体外和遗传操作,以确定潜在的 CypD 磷酸化位点及其对线粒体功能和细胞死亡的影响。重要的是,我们开发了一种体内心肌内腺病毒策略来评估 CypD 磷酸化事件对梗死面积的影响。我们的结果表明,尽管 CypD 可以潜在地在多个丝氨酸残基上被磷酸化,但只有 S191 位点的磷酸化状态直接影响 CypD 调节 mPTP 的能力。蛋白质-蛋白质相互作用策略表明,在耐磷酸化的 S191A 突变体中,CypD 与寡霉素敏感性 conferring protein (OSCP) 的相互作用降低了 45%,而在磷酸化模拟的 S191E 突变体细胞中则增加了 48%。结果,耐磷酸化的 CypD S191A 突变体在 18 小时饥饿中得到保护,而磷酸化模拟的 S191E 组的细胞死亡明显增加,伴随着线粒体呼吸改变和 ROS 产生。作为体内概念验证,在 S191A 耐磷酸化的 CypD-KO 小鼠中,梗死面积明显小于 WT,而在 S191E 磷酸化模拟的 CypD-KO 小鼠中,梗死面积则明显增加。我们得出结论,CypD 在 S191 残基上的磷酸化导致其与 OSCP 结合,从而使 mPTP 对随后的细胞死亡敏感。