Shanghai Institute of Stem Cell Research and Clinical Translation, Shanghai East Hospital, Tongji University School of Medicine, Shanghai, 200120, China.
State Key Laboratory of Medical Neurobiology and MOE Frontiers Center for Brain Science, Institutes of Brain Science, Jing'an District Centre Hospital of Shanghai, Fudan University, Shanghai, 200032, China.
Protein Cell. 2021 Aug;12(8):639-652. doi: 10.1007/s13238-020-00773-z. Epub 2020 Aug 27.
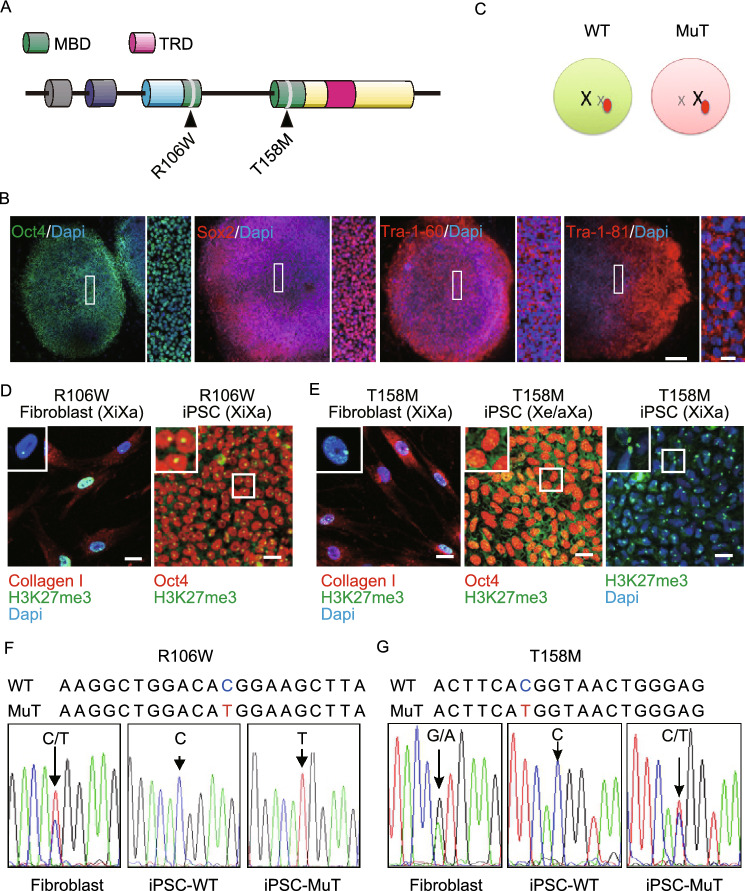
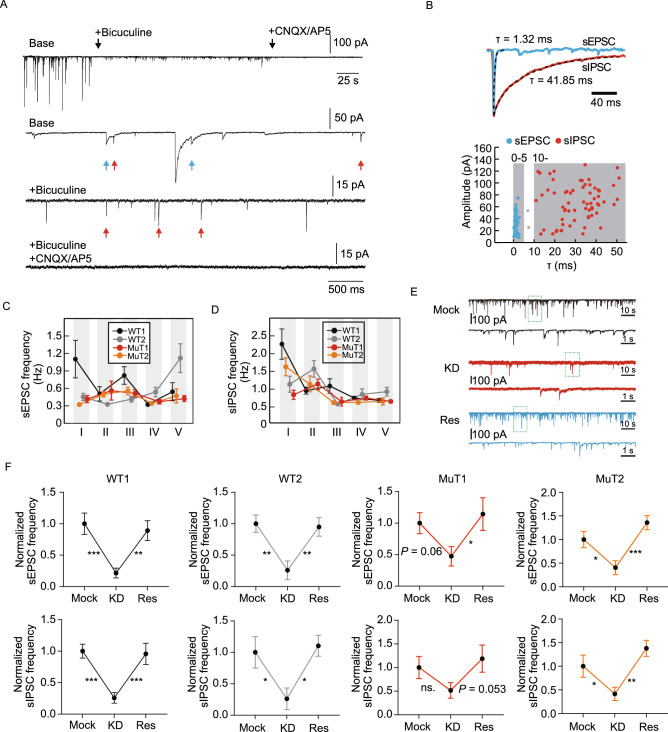
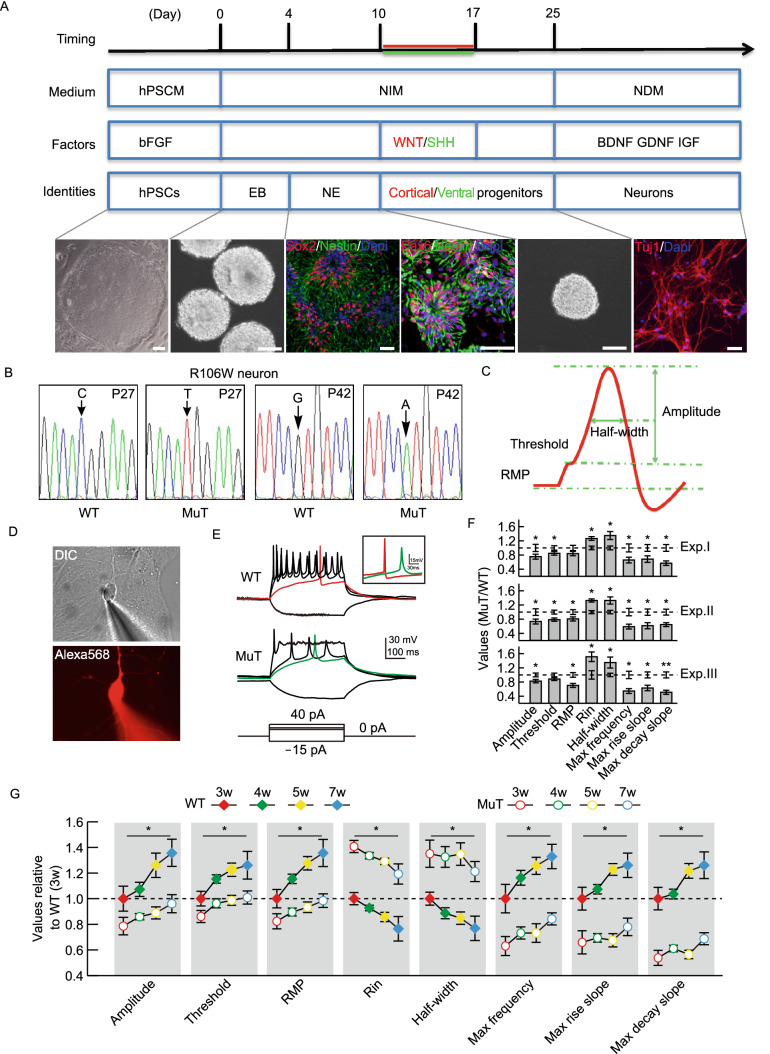
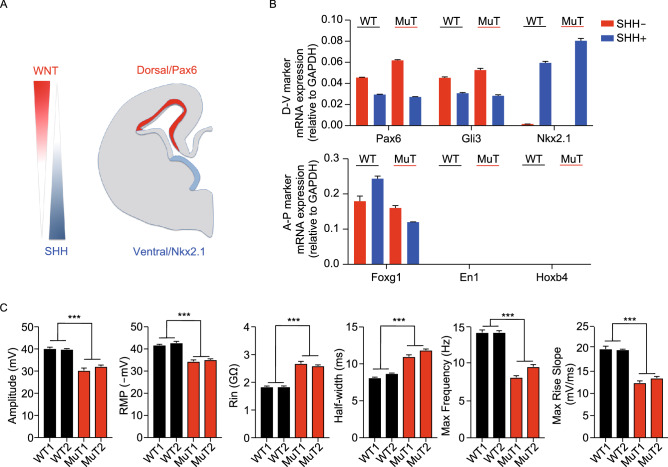
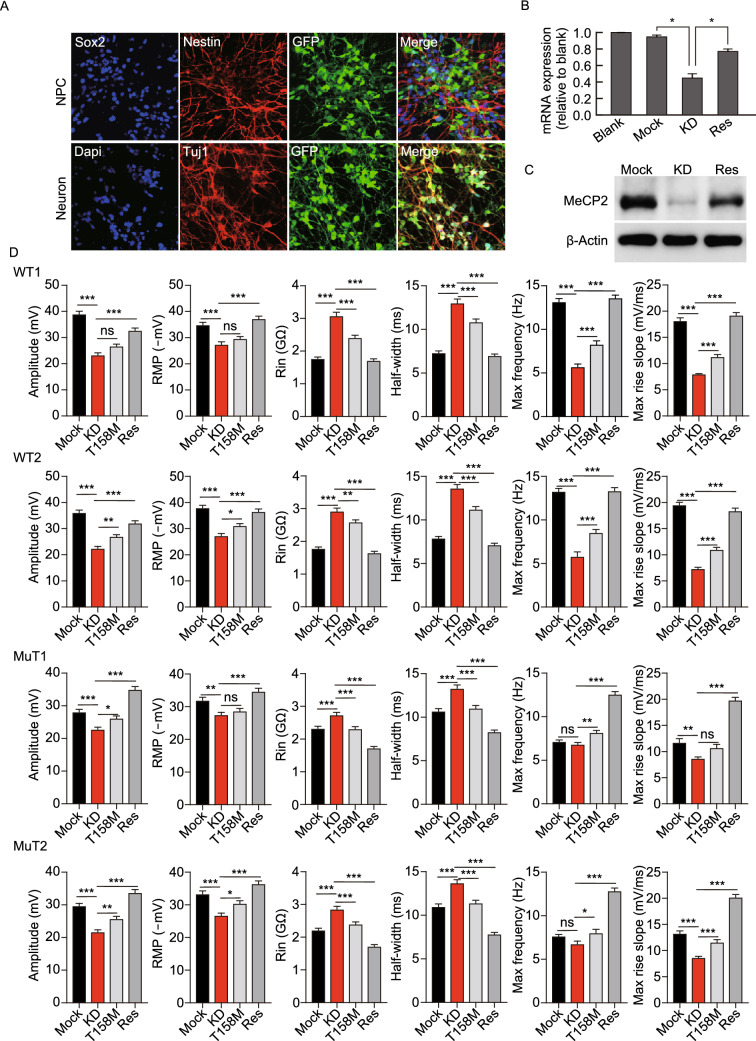
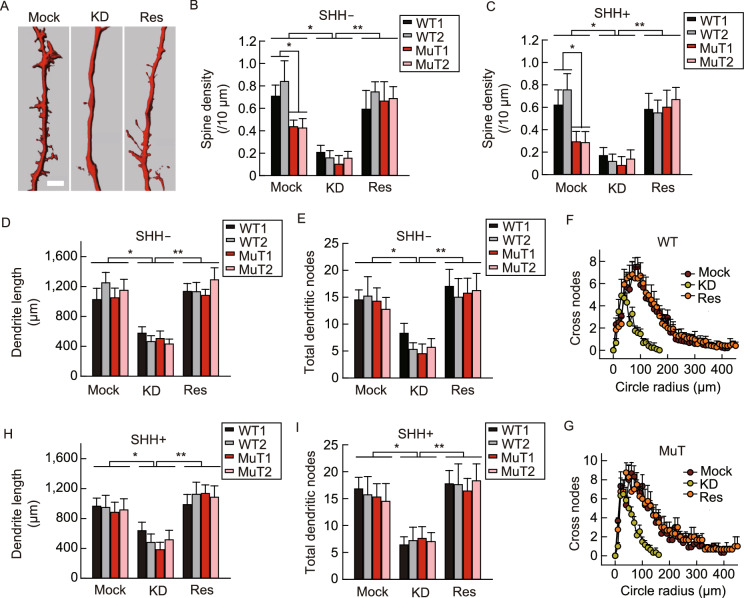
Rett syndrome (RTT) is a progressive neurodevelopmental disorder, mainly caused by mutations in MeCP2 and currently with no cure. We report here that neurons from R106W MeCP2 RTT human iPSCs as well as human embryonic stem cells after MeCP2 knockdown exhibit consistent and long-lasting impairment in maturation as indicated by impaired action potentials and passive membrane properties as well as reduced soma size and spine density. Moreover, RTT-inherent defects in neuronal maturation could be pan-neuronal and occurred in neurons with both dorsal and ventral forebrain features. Knockdown of MeCP2 led to more severe neuronal deficits as compared to RTT iPSC-derived neurons, which appeared to retain partial function. Strikingly, consistent deficits in nuclear size, dendritic complexity and circuitry-dependent spontaneous postsynaptic currents could only be observed in MeCP2 knockdown neurons but not RTT iPSC-derived neurons. Both neuron-intrinsic and circuitry-dependent deficits of MeCP2-deficient neurons could be fully or partially rescued by re-expression of wild type or T158M MeCP2, strengthening the dosage dependency of MeCP2 on disease phenotypes and also the partial function of the mutant. Our findings thus reveal stable neuronal maturation deficits and unexpectedly, graded sensitivities of neuron-inherent and neural transmission phenotypes towards the extent of MeCP2 deficiency, which is informative for future therapeutic development.
雷特综合征(RTT)是一种进行性神经发育障碍,主要由 MeCP2 基因突变引起,目前尚无治愈方法。我们在这里报告,R106W MeCP2 RTT 人诱导多能干细胞以及 MeCP2 敲低后的人胚胎干细胞中的神经元表现出一致且持久的成熟障碍,表现为动作电位和被动膜特性受损,以及胞体大小和棘突密度降低。此外,神经元成熟的 RTT 固有缺陷可能是全神经元的,并且发生在具有背侧和腹侧前脑特征的神经元中。与 RTT 诱导多能干细胞衍生的神经元相比,MeCP2 敲低导致更严重的神经元缺陷,这些神经元似乎保留了部分功能。引人注目的是,只有在 MeCP2 敲低神经元中才能观察到核大小、树突复杂性和与电路相关的自发性突触后电流的一致缺陷,而在 RTT 诱导多能干细胞衍生的神经元中则没有。MeCP2 缺失神经元的内在神经元和电路依赖性缺陷均可通过野生型或 T158M MeCP2 的重新表达得到完全或部分挽救,这增强了 MeCP2 对疾病表型的剂量依赖性,以及突变体的部分功能。因此,我们的研究结果揭示了稳定的神经元成熟缺陷,出乎意料的是,内在神经元和神经传递表型对 MeCP2 缺乏程度的敏感性存在分级差异,这对未来的治疗开发具有重要意义。