Department of Molecular Genetics, University of Toronto, Toronto, ON, M5S 1A8, Canada.
Developmental & Stem Cell Biology Program, The Hospital for Sick Children, Toronto, ON, M5G 0A4, Canada.
Transl Psychiatry. 2022 Oct 18;12(1):450. doi: 10.1038/s41398-022-02216-1.
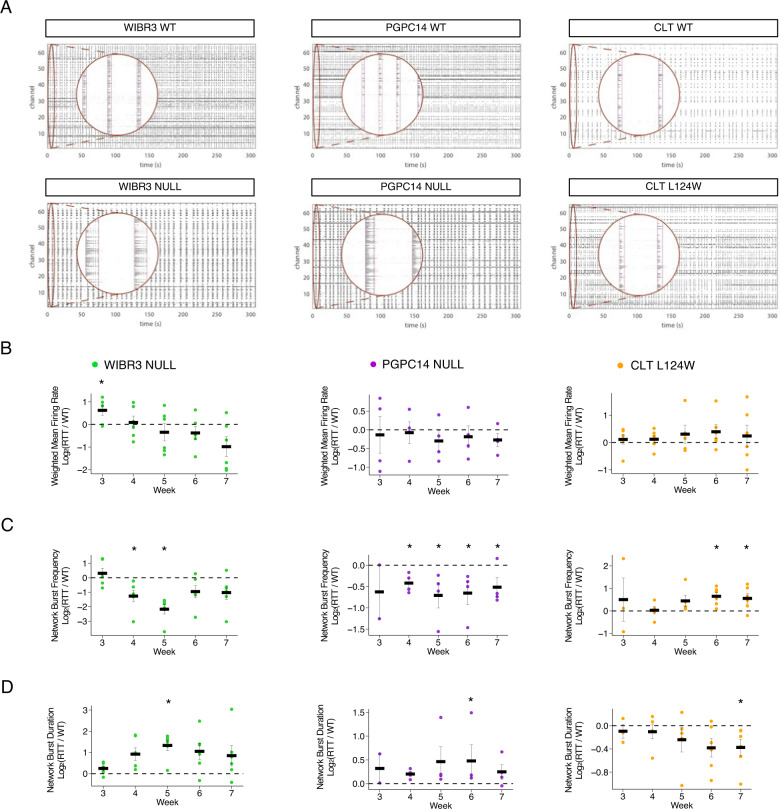
Rett syndrome (RTT) is a severe neurodevelopmental disorder primarily caused by heterozygous loss-of-function mutations in the X-linked gene MECP2 that is a global transcriptional regulator. Mutations in the methyl-CpG binding domain (MBD) of MECP2 disrupt its interaction with methylated DNA. Here, we investigate the effect of a novel MECP2 L124W missense mutation in the MBD of an atypical RTT patient with preserved speech in comparison to severe MECP2 null mutations. L124W protein had a limited ability to disrupt heterochromatic chromocenters due to decreased binding dynamics. We isolated two pairs of isogenic WT and L124W induced pluripotent stem cells. L124W induced excitatory neurons expressed stable protein, exhibited increased input resistance and decreased voltage-gated Na and K currents, and their neuronal dysmorphology was limited to decreased dendritic complexity. Three isogenic pairs of MECP2 null neurons had the expected more extreme morphological and electrophysiological phenotypes. We examined development and maturation of L124W and MECP2 null excitatory neural network activity using micro-electrode arrays. Relative to isogenic controls, L124W neurons had an increase in synchronous network burst frequency, in contrast to MECP2 null neurons that suffered a significant decrease in synchronous network burst frequency and a transient extension of network burst duration. A biologically motivated computational neural network model shows the observed changes in network dynamics are explained by changes in intrinsic Na and K currents in individual neurons. Our multilevel results demonstrate that RTT excitatory neurons show a wide spectrum of morphological, electrophysiological and circuitry phenotypes that are dependent on the severity of the MECP2 mutation.
雷特综合征(RTT)是一种严重的神经发育障碍,主要由 X 连锁基因 MECP2 的杂合功能丧失突变引起,该基因是一种全局转录调节剂。MECP2 中的甲基-CpG 结合结构域(MBD)突变会破坏其与甲基化 DNA 的相互作用。在这里,我们研究了一种新型 MECP2 L124W 错义突变在具有保留言语的非典型 RTT 患者中的作用,与严重的 MECP2 缺失突变相比。由于结合动力学降低,L124W 蛋白破坏异染色质染色体中心的能力有限。我们分离了两对同基因 WT 和 L124W 诱导多能干细胞。L124W 诱导的兴奋性神经元表达稳定的蛋白质,表现出增加的输入电阻和减少的电压门控 Na 和 K 电流,并且它们的神经元形态异常仅限于减少树突复杂性。三对同基因 MECP2 缺失神经元具有预期的更极端的形态和电生理表型。我们使用微电极阵列检查 L124W 和 MECP2 缺失兴奋性神经网络活动的发育和成熟。与同基因对照相比,L124W 神经元的同步网络爆发频率增加,而 MECP2 缺失神经元的同步网络爆发频率显著降低,网络爆发持续时间短暂延长。具有生物学意义的计算神经网络模型表明,观察到的网络动力学变化可以通过单个神经元中的内在 Na 和 K 电流变化来解释。我们的多层次结果表明,RTT 兴奋性神经元表现出广泛的形态、电生理和电路表型,这些表型取决于 MECP2 突变的严重程度。