Ricardi Niccolò, Ernst Michelle, Macchi Piero, Wesolowski Tomasz Adam
Department of Physical Chemistry, University of Geneva, 30, Quai Ernest-Ansermet, CH-1211 Genève 4, Switzerland.
University of Bern, Freiestraße 3, 3012 Bern, Switzerland.
Acta Crystallogr A Found Adv. 2020 Sep 1;76(Pt 5):571-579. doi: 10.1107/S2053273320008062. Epub 2020 Jul 20.
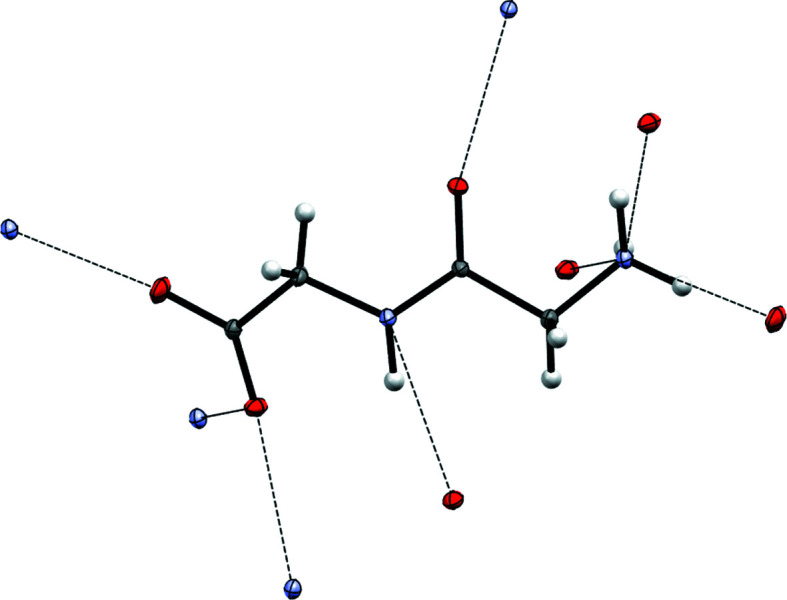
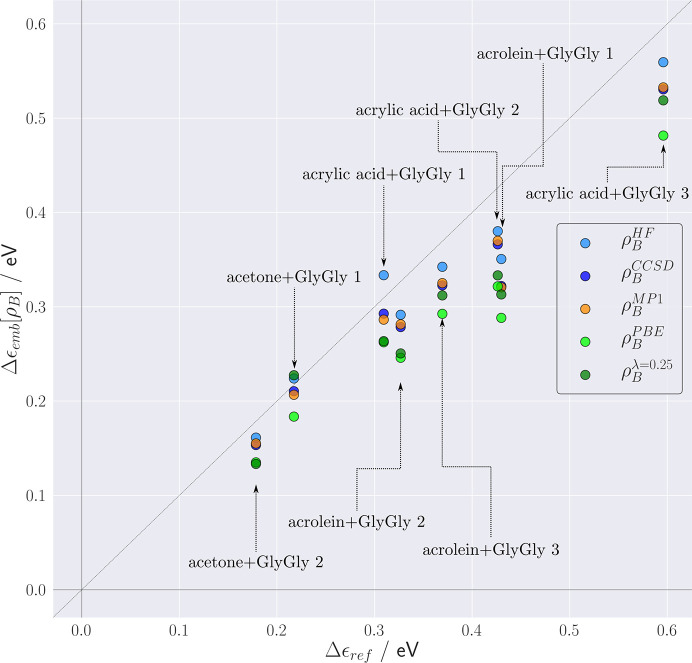
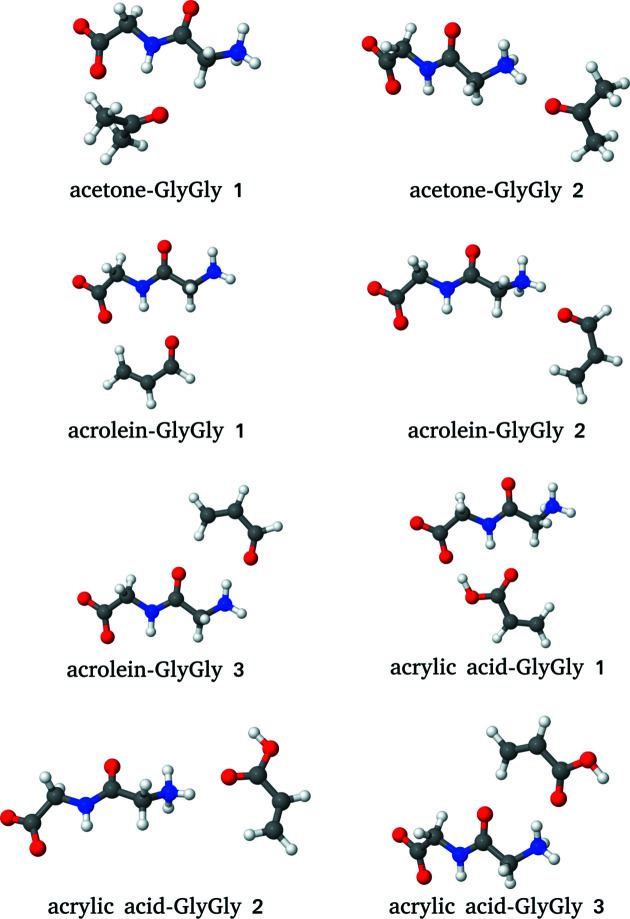
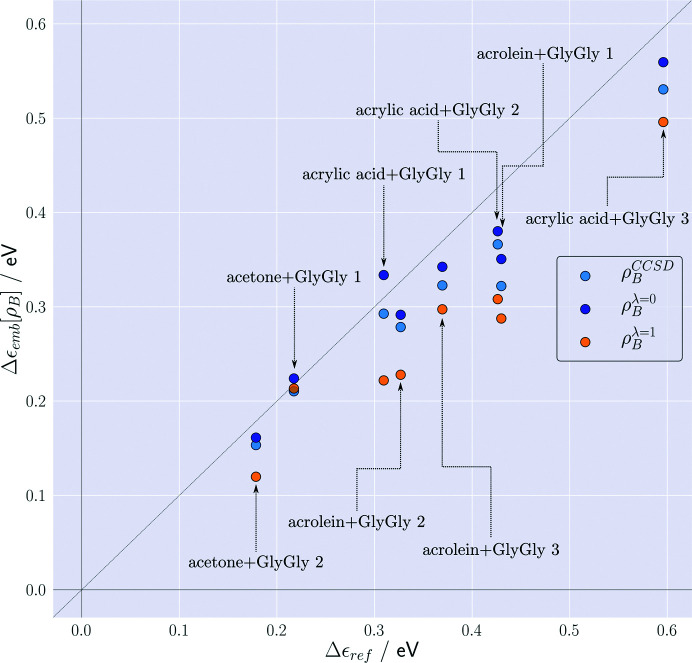
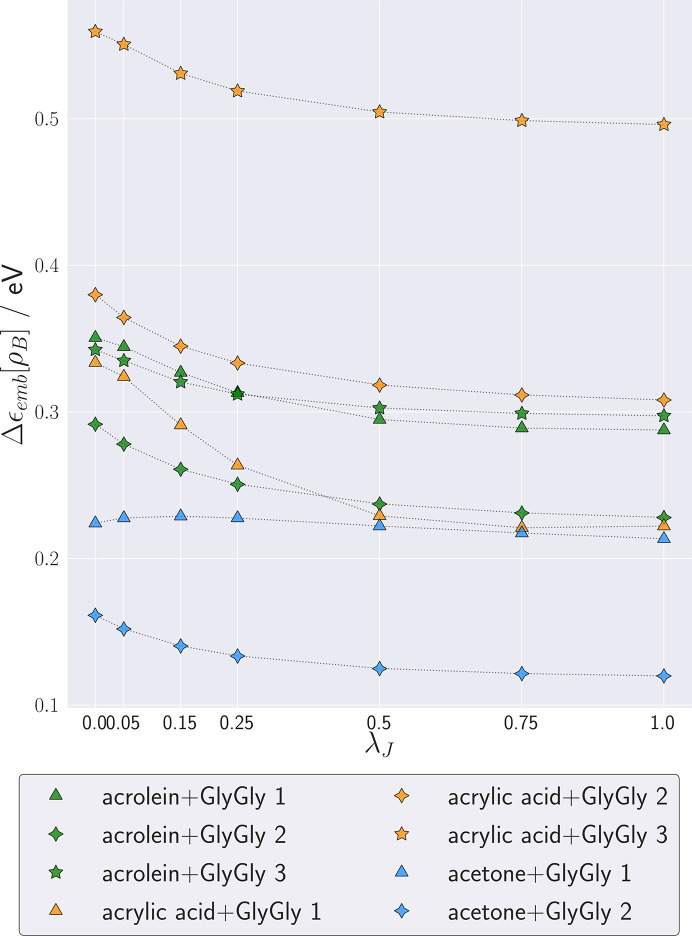
The basic idea of frozen-density embedding theory (FDET) is the constrained minimization of the Hohenberg-Kohn density functional E[ρ] performed using the auxiliary functional E_{v_{AB}}^{\rm FDET}[\Psi A, \rho B], where Ψ is the embedded N-electron wavefunction and ρ(r) is a non-negative function in real space integrating to a given number of electrons N. This choice of independent variables in the total energy functional E{v{AB}}^{\rm FDET}[\Psi _A, \rho _B] makes it possible to treat the corresponding two components of the total density using different methods in multi-level simulations. The application of FDET using ρ(r) reconstructed from X-ray diffraction data for a molecular crystal is demonstrated for the first time. For eight hydrogen-bonded clusters involving a chromophore (represented as Ψ) and the glycylglycine molecule [represented as ρ(r)], FDET is used to derive excitation energies. It is shown that experimental densities are suitable for use as ρ(r) in FDET-based simulations.
冻结密度嵌入理论(FDET)的基本思想是利用辅助泛函(E_{v_{AB}}^{\rm FDET}[\Psi A, \rho B])对 Hohenberg-Kohn 密度泛函(E[ρ])进行约束最小化,其中(\Psi)是嵌入的(N)电子波函数,(\rho(r))是实空间中积分为给定电子数(N)的非负函数。总能量泛函(E{v{AB}}^{\rm FDET}[\Psi _A, \rho _B])中这种自变量的选择使得在多级模拟中可以使用不同方法处理总密度的相应两个分量。首次展示了将 FDET 应用于由分子晶体的 X 射线衍射数据重建的(\rho(r))。对于八个涉及发色团(表示为(\Psi))和甘氨酰甘氨酸分子[表示为(\rho(r))]的氢键簇,使用 FDET 推导激发能。结果表明,实验密度适用于基于 FDET 的模拟中的(\rho(r))。