Department of Biology, University of North Carolina Greensboro, Greensboro, NC.
Mol Biol Evol. 2021 Jan 23;38(2):727-734. doi: 10.1093/molbev/msaa224.
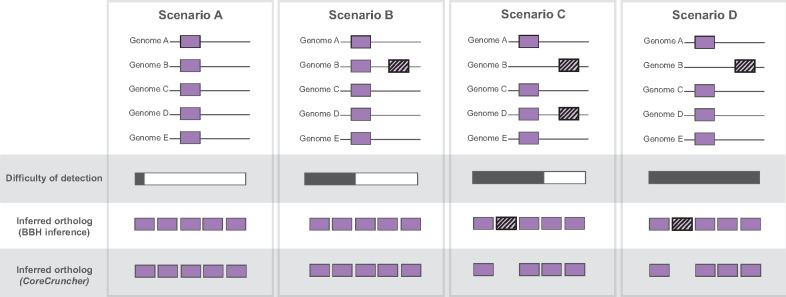
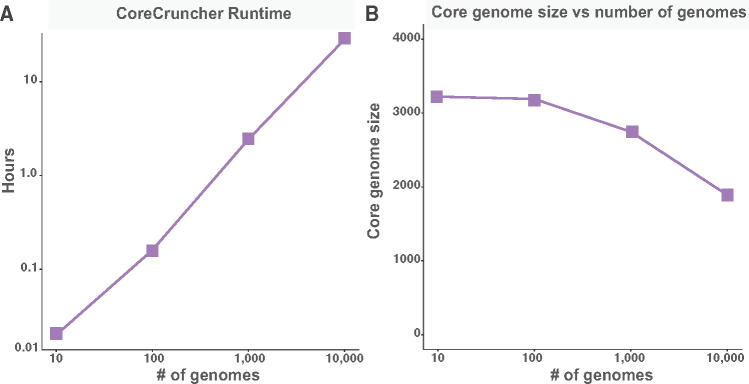
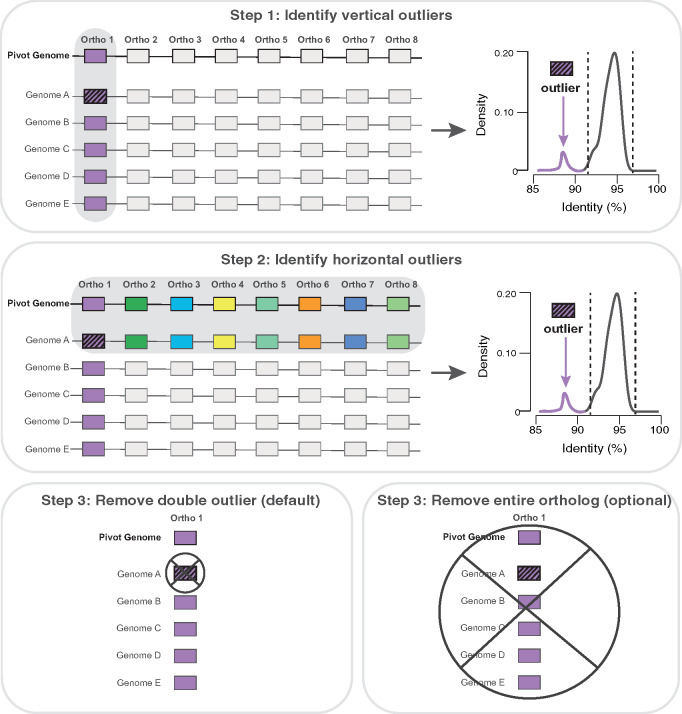
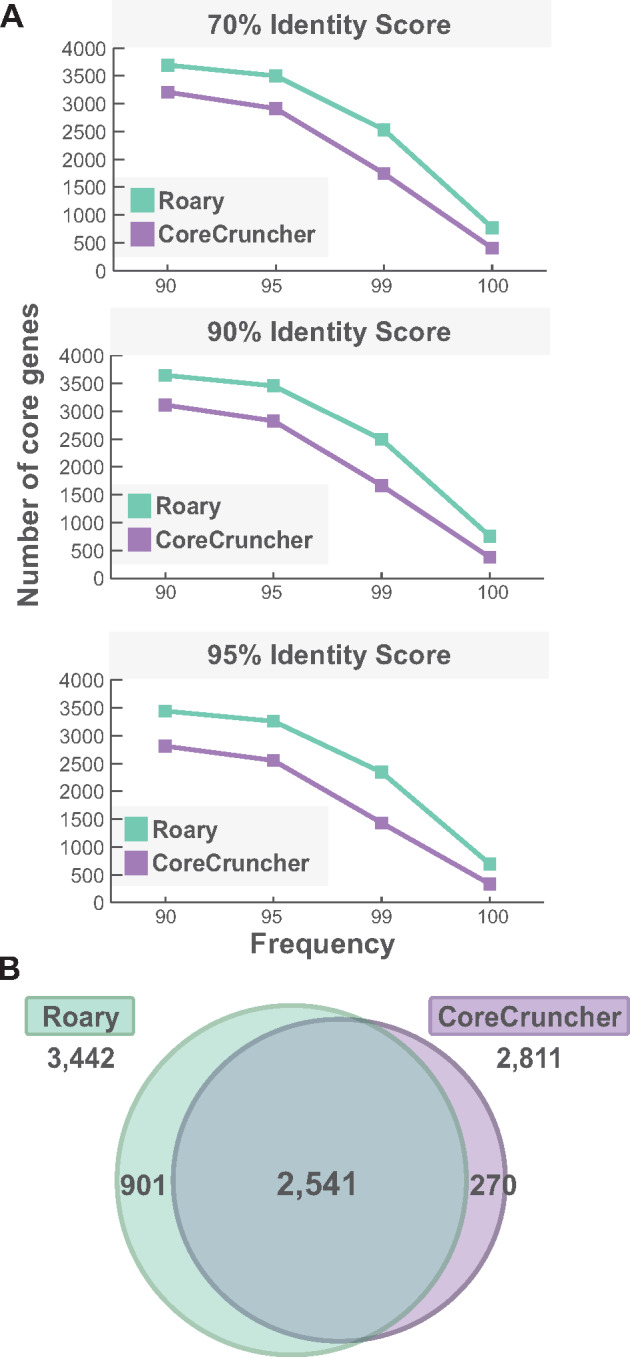
The core genome represents the set of genes shared by all, or nearly all, strains of a given population or species of prokaryotes. Inferring the core genome is integral to many genomic analyses, however, most methods rely on the comparison of all the pairs of genomes; a step that is becoming increasingly difficult given the massive accumulation of genomic data. Here, we present CoreCruncher; a program that robustly and rapidly constructs core genomes across hundreds or thousands of genomes. CoreCruncher does not compute all pairwise genome comparisons and uses a heuristic based on the distributions of identity scores to classify sequences as orthologs or paralogs/xenologs. Although it is much faster than current methods, our results indicate that our approach is more conservative than other tools and less sensitive to the presence of paralogs and xenologs. CoreCruncher is freely available from: https://github.com/lbobay/CoreCruncher. CoreCruncher is written in Python 3.7 and can also run on Python 2.7 without modification. It requires the python library Numpy and either Usearch or Blast. Certain options require the programs muscle or mafft.
核心基因组代表了给定种群或原核生物物种中所有或几乎所有菌株所共有的基因集。推断核心基因组是许多基因组分析的重要组成部分,然而,大多数方法都依赖于所有基因组对的比较;随着基因组数据的大量积累,这一步变得越来越困难。在这里,我们介绍 CoreCruncher;这是一个程序,可以快速稳健地构建数百或数千个基因组的核心基因组。CoreCruncher 不会计算所有的成对基因组比较,而是使用基于身份分数分布的启发式方法将序列分类为直系同源物或旁系同源物/异源同源物。虽然它比当前的方法快得多,但我们的结果表明,我们的方法比其他工具更保守,对旁系同源物和异源同源物的存在不太敏感。CoreCruncher 可从以下网址免费获得:https://github.com/lbobay/CoreCruncher。CoreCruncher 是用 Python 3.7 编写的,也可以在不修改的情况下在 Python 2.7 上运行。它需要 Python 库 Numpy 和 Usearch 或 Blast。某些选项需要 muscle 或 mafft 程序。