Department of Animal Genetics, Breeding and Reproduction, College of Animal Science and Technology, Hebei Agricultural University, Lekai South Street No. 2596, Baoding, 071000, Hebei, China.
Engineering Research Center for Agriculture in Hebei Mountainous Areas, Baoding, 071000, Hebei, China.
BMC Genomics. 2020 Sep 14;21(1):636. doi: 10.1186/s12864-020-07055-2.
Improving sow fertility is extremely important as it can lead to increased reproductive efficiency and thus profitability for swine producers. There are considerable differences in fertility rates among individual animals, but the underlying molecular mechanisms remain unclear. In this study, by using different types of RNA libraries, we investigated the complete transcriptome of ovarian tissue during the luteal (L) and follicular (F) phases of the estrous cycle in Large White pigs with high (H) and low (L) fecundity, and performed a comprehensive analysis of long noncoding RNAs (lncRNAs), mRNAs and micro RNAs (miRNAs) from 16 samples by combining RNA sequencing (RNA-seq) with bioinformatics.
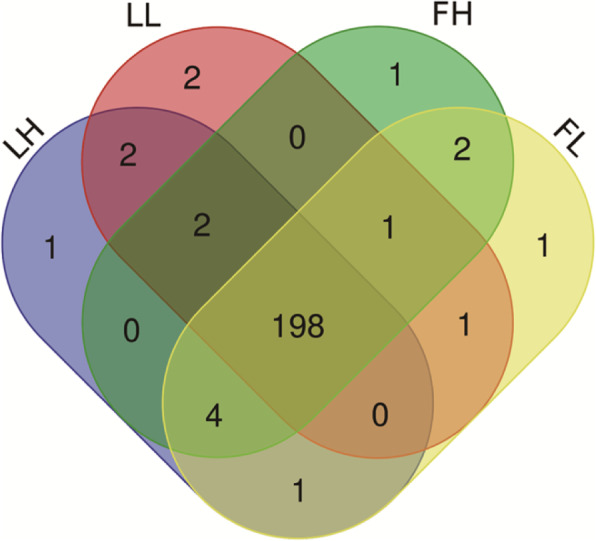
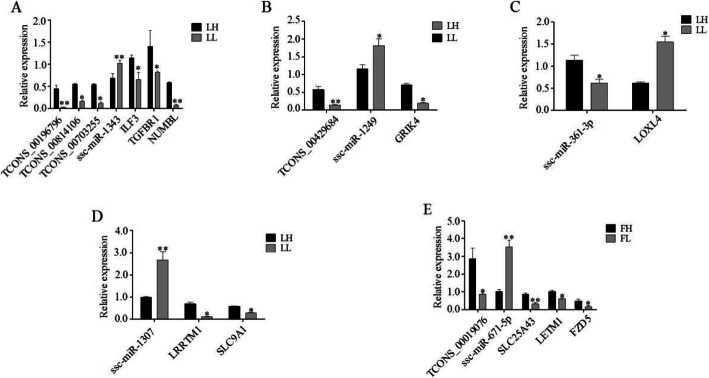
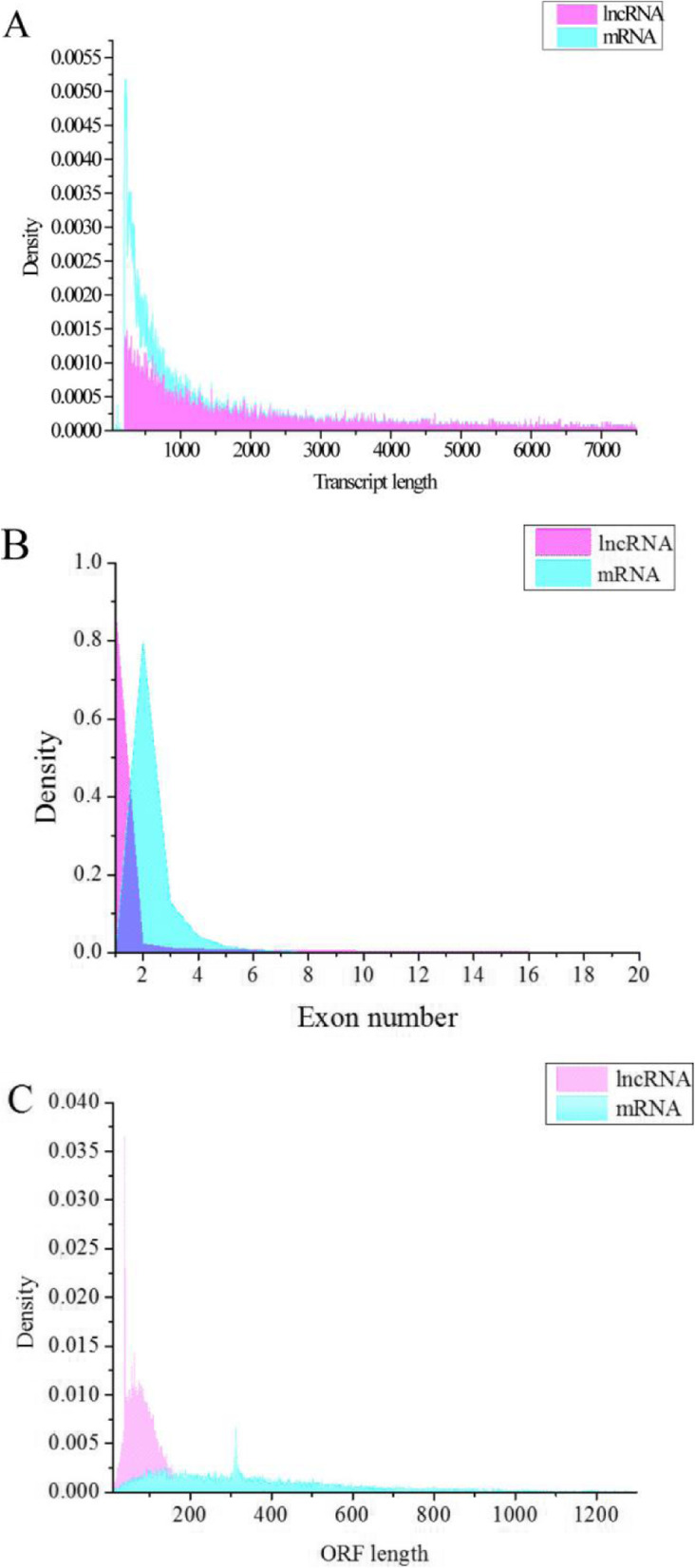
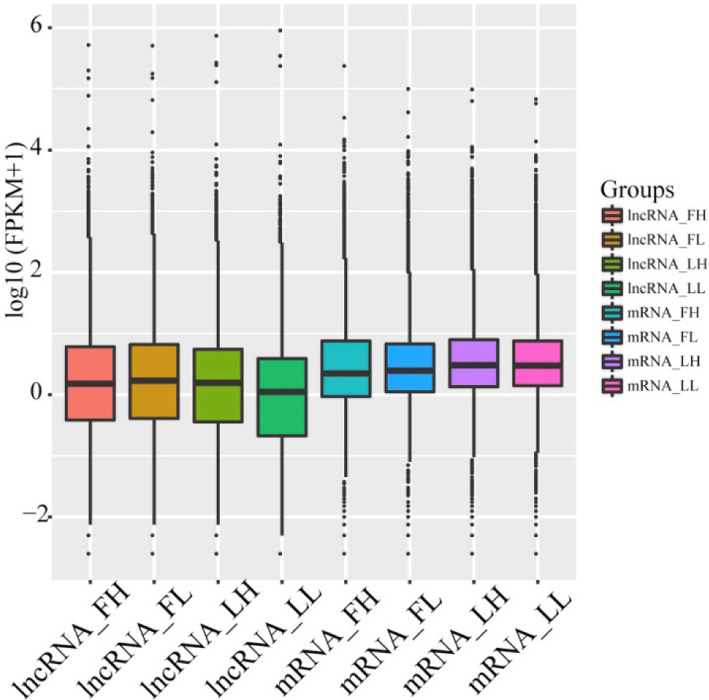
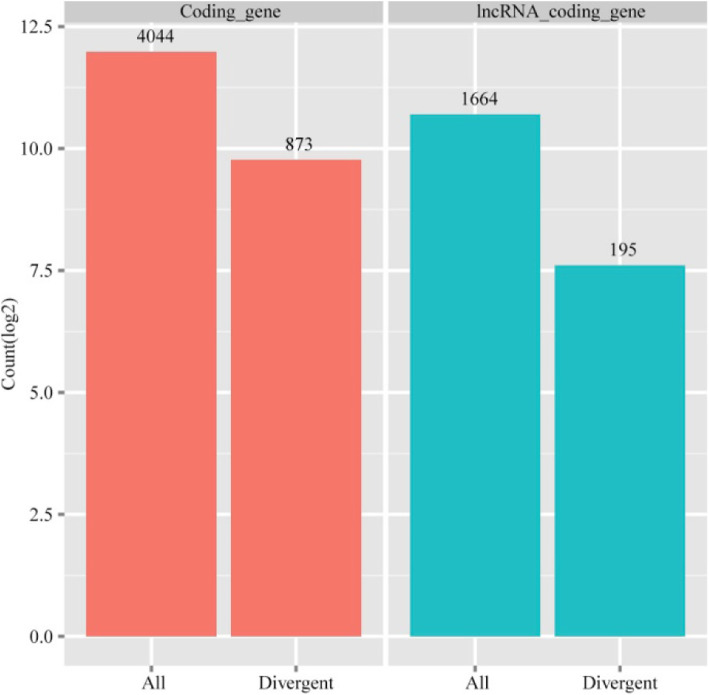
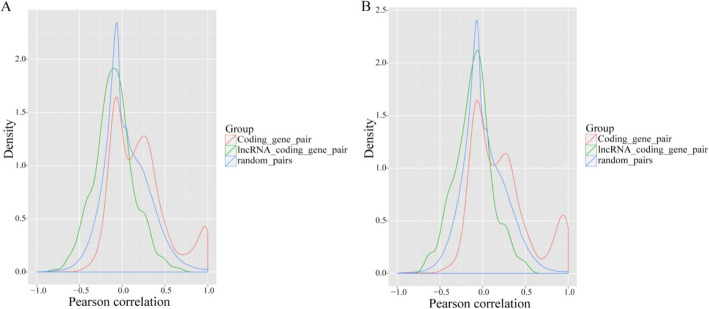
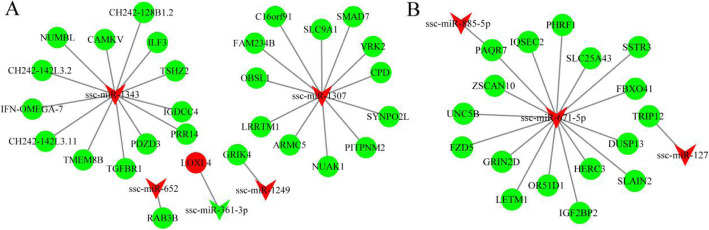
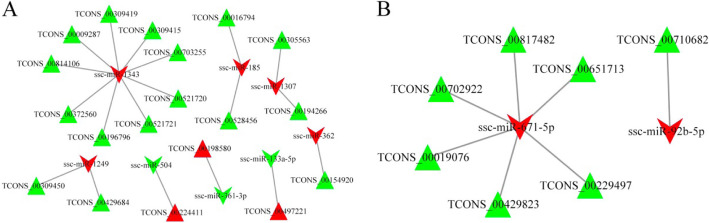
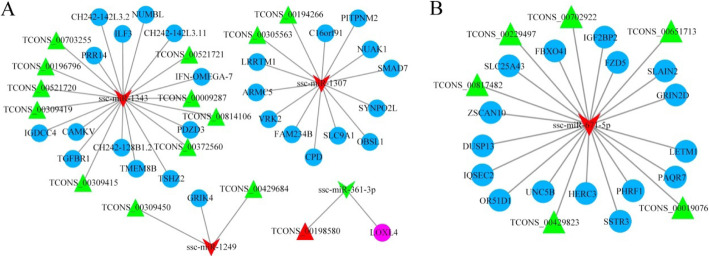
In total, 24,447 lncRNAs, 27,370 mRNAs, and 216 known miRNAs were identified in ovarian tissues. The genomic features of lncRNAs, such as length distribution and number of exons, were further analyzed. We selected a threshold of P < 0.05 and |log (fold change)| ≥ 1 to obtain the differentially expressed lncRNAs, miRNAs and mRNAs by pairwise comparison (LH vs. LL, FH vs. FL). Bioinformatics analysis of these differentially expressed RNAs revealed multiple significantly enriched pathways (P < 0.05) that were closely involved in the reproductive process, such as ovarian steroidogenesis, lysosome, steroid biosynthesis, and the estrogen and GnRH signaling pathways. Moreover, bioinformatics screening of differentially expressed miRNAs that share common miRNA response elements (MREs) with lncRNAs and their downstream mRNA targets were performed. Finally, we constructed lncRNA-miRNA-mRNA regulation networks. The key genes in these networks were verified by Reverse Transcription Real-time Quantitative PCR (RT-qRCR), which were consistent with the results from RNA-Seq data.
These results provide further insights into the fertility of pigs andcan contribute to further experimental investigation of the functions of these genes.
提高母猪的繁殖力极为重要,因为这可以提高养猪生产者的繁殖效率,从而提高盈利能力。个体动物之间的繁殖力存在很大差异,但潜在的分子机制尚不清楚。在这项研究中,我们使用不同类型的 RNA 文库,研究了高(H)和低(L)产力大白猪发情周期黄体(L)和卵泡(F)阶段卵巢组织的完整转录组,并通过将 RNA 测序(RNA-seq)与生物信息学相结合,对 16 个样本中的长非编码 RNA(lncRNA)、mRNA 和 microRNA(miRNA)进行了全面分析。
共鉴定出卵巢组织中的 24447 个 lncRNA、27370 个 mRNA 和 216 个已知 miRNA。进一步分析了 lncRNA 的基因组特征,如长度分布和外显子数量。我们通过成对比较(LH 与 LL、FH 与 FL)选择 P<0.05 和|log(fold change)|≥1 作为差异表达 lncRNA、miRNA 和 mRNA 的阈值。对这些差异表达 RNA 的生物信息学分析揭示了多个与生殖过程密切相关的显著富集途径(P<0.05),如卵巢甾体生成、溶酶体、甾体生物合成以及雌激素和 GnRH 信号通路。此外,对与 lncRNA 及其下游 mRNA 靶标共享共同 miRNA 反应元件(MRE)的差异表达 miRNA 进行了生物信息学筛选。最后,我们构建了 lncRNA-miRNA-mRNA 调控网络。通过逆转录实时定量 PCR(RT-qRCR)验证了这些网络中的关键基因,结果与 RNA-seq 数据一致。
这些结果为猪的繁殖力提供了进一步的认识,并有助于进一步实验研究这些基因的功能。