Graduate Program in Innovation Pharmaceutical, Federal University of Amapá, 68903-419 Amapá-AP, Brazil.
Laboratory of Modeling and Computational Chemistry, Department of Biological and Health Sciences, Federal University of Amapá, 68902-280 Macapá-AP, Brazil.
Molecules. 2020 Sep 12;25(18):4183. doi: 10.3390/molecules25184183.
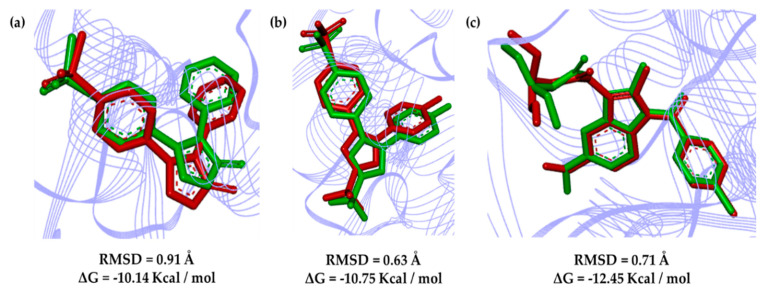
Non-steroidal anti-inflammatory drugs are inhibitors of cyclooxygenase-2 (COX-2) that were developed in order to avoid the side effects of non-selective inhibitors of COX-1. Thus, the present study aims to identify new selective chemical entities for the COX-2 enzyme via molecular modeling approaches. The best pharmacophore model was used to identify compounds within the ZINC database. The molecular properties were determined and selected with Pearson's correlation for the construction of quantitative structure-activity relationship (QSAR) models to predict the biological activities of the compounds obtained with virtual screening. The pharmacokinetic/toxicological profiles of the compounds were determined, as well as the binding modes through molecular docking compared to commercial compounds (rofecoxib and celecoxib). The QSAR analysis showed a fit with R = 0.9617, R = 0.9250, standard error of estimate (SEE) = 0.2238, and F = 46.2739, with the tetra-parametric regression model. After the analysis, only three promising inhibitors were selected, , , and , with their predicted pIC (-log IC) values, = 7.9484, = 9.3458, and = 9.5272. All candidates inhibitors complied with Lipinski's rule of five, which predicts a good oral availability and can be used in in vitro and in vivo tests in the zebrafish model in order to confirm the obtained in silico data.
非甾体抗炎药是环氧化酶-2(COX-2)的抑制剂,为了避免 COX-1 非选择性抑制剂的副作用而开发。因此,本研究旨在通过分子建模方法为 COX-2 酶鉴定新的选择性化学实体。最佳药效团模型用于从 ZINC 数据库中识别化合物。通过皮尔逊相关性确定和选择分子特性,以构建定量构效关系(QSAR)模型,预测虚拟筛选获得的化合物的生物活性。测定了化合物的药代动力学/毒理学特征,并通过与商业化合物(罗非昔布和塞来昔布)的分子对接比较确定了结合模式。QSAR 分析显示拟合度良好,R = 0.9617,R = 0.9250,估计标准误差(SEE)= 0.2238,F = 46.2739,采用四参数回归模型。分析后,仅选择了三种有前途的抑制剂, , 和 ,其预测的 pIC(-log IC)值分别为 = 7.9484, = 9.3458, = 9.5272。所有候选抑制剂都符合 Lipinski 的五规则,这表明它们具有良好的口服生物利用度,可以在体外和体内斑马鱼模型中进行测试,以确认获得的计算机数据。