Tsuboi Yoshio, Mishima Takayasu, Fujioka Shinsuke
Department of Neurology, School of Medicine, Fukuoka University, Fukuoka, Japan.
J Mov Disord. 2021 Jan;14(1):1-9. doi: 10.14802/jmd.20060. Epub 2020 Sep 21.
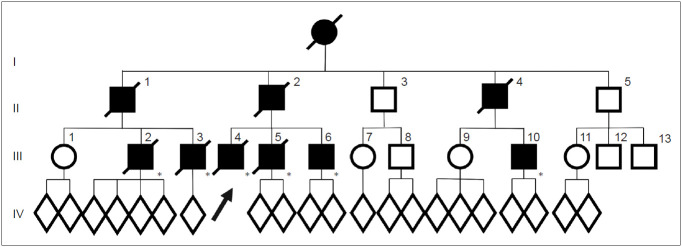
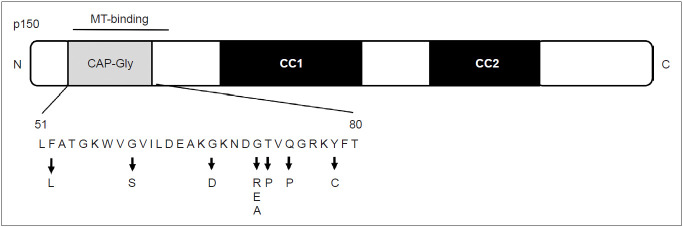
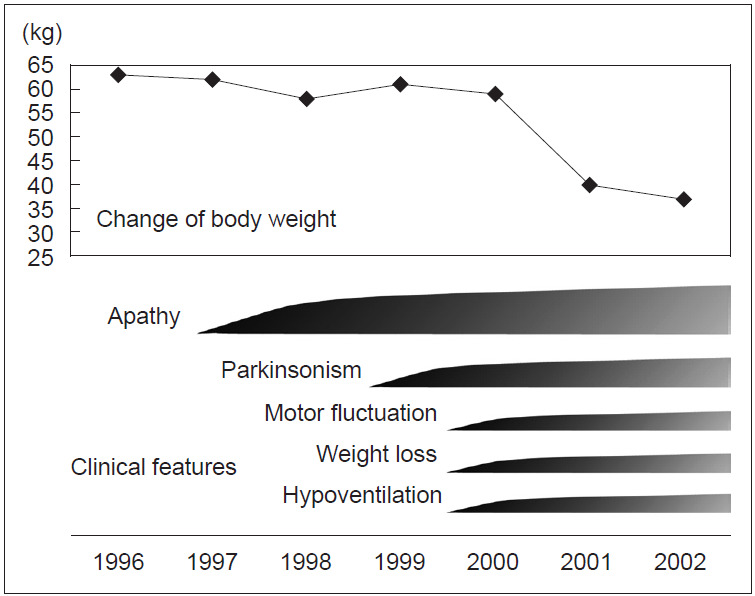
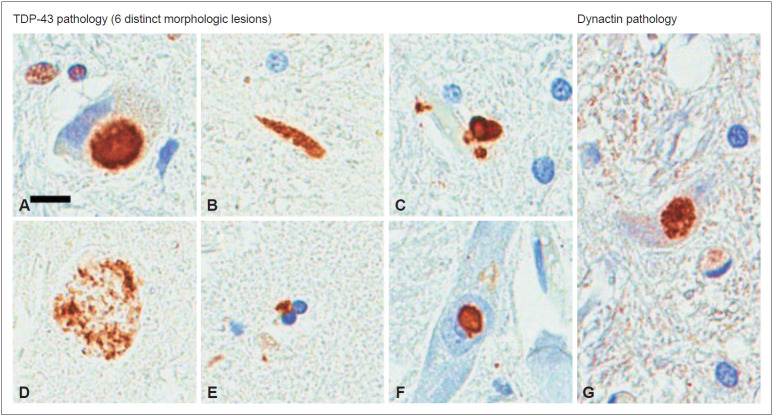
Perry disease is a hereditary neurodegenerative disease with autosomal dominant inheritance. It is characterized by parkinsonism, psychiatric symptoms, unexpected weight loss, central hypoventilation, and transactive-response DNA-binding protein of 43kD (TDP-43) aggregation in the brain. In 2009, Perry disease was found to be caused by dynactin I gene (DCTN1), which encodes dynactin subunit p150 on chromosome 2p, in patients with the disease. The dynactin complex is a motor protein that is associated with axonal transport. Presently, at least 8 mutations and 22 families have been reported; other than the "classic" syndrome, distinct phenotypes are recognized. The neuropathology of Perry disease reveals severe degeneration in the substantia nigra and TDP-43 inclusions in the basal ganglia and brain stem. How dysfunction of the dynactin molecule is related to TDP-43 pathology in Perry disease is important to elucidate the pathological mechanism and develop new treatment.
佩里病是一种常染色体显性遗传的遗传性神经退行性疾病。其特征包括帕金森综合征、精神症状、意外体重减轻、中枢性通气不足以及大脑中43kD的反式激活反应DNA结合蛋白(TDP - 43)聚集。2009年,在佩里病患者中发现该病由动力蛋白激活蛋白I基因(DCTN1)引起,该基因在2号染色体短臂上编码动力蛋白激活蛋白亚基p150。动力蛋白激活蛋白复合体是一种与轴突运输相关的运动蛋白。目前,已报道至少8种突变和22个家系;除了“经典”综合征外,还识别出了不同的表型。佩里病的神经病理学显示黑质严重退化,基底神经节和脑干中有TDP - 43包涵体。阐明动力蛋白分子功能障碍与佩里病中TDP - 43病理学如何相关,对于阐明病理机制和开发新的治疗方法很重要。