Noman Kinza, Hendriksz Christian J, Radcliffe Graham, Roncaroli Federico, Moreea Sulleman, Hussain Afifah, Stepien Karolina M
Medical School, University of Manchester, United Kingdom.
University of Pretoria, Steve Biko Academic Unit, Paediatrics and Child Health, Pretoria, South Africa.
Mol Genet Metab Rep. 2020 Sep 7;25:100646. doi: 10.1016/j.ymgmr.2020.100646. eCollection 2020 Dec.
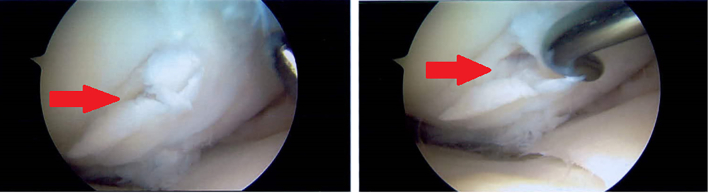
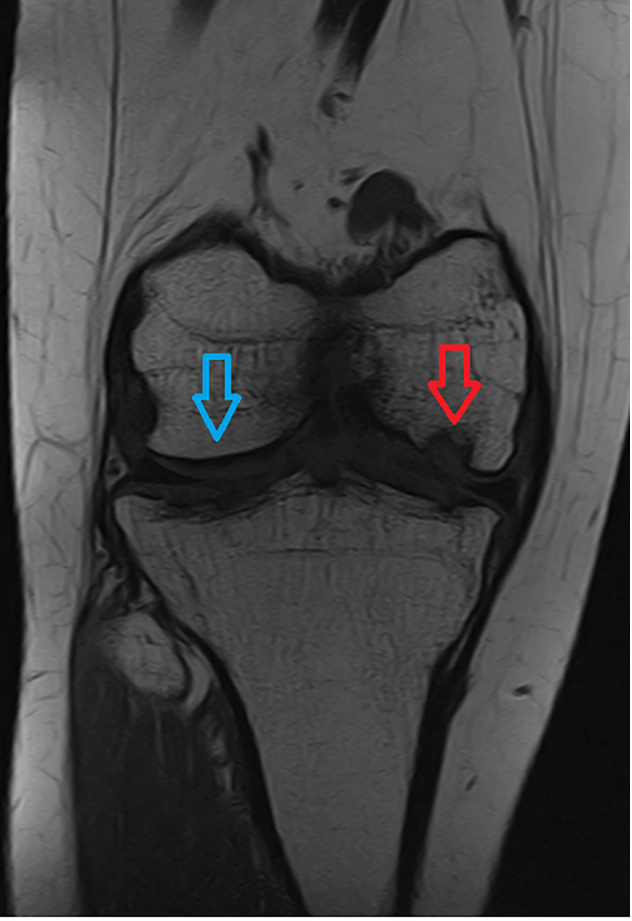
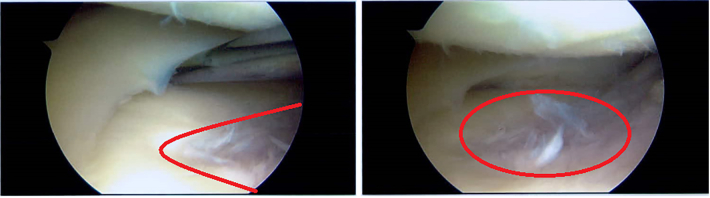
The mannose phosphate isomerase-congenital disorder of glycosylation (MPI-CDG) is caused by phosphomannose isomerase deficiency. Clinical features include hyperinsulinaemic hypoglycaemia, protein losing enteropathy, hepatomegaly and hepatic fibrosis, digestive symptoms and coagulation abnormalities. The condition is treated with mannose supplementation. Long-term outcomes in adults are not well described. We present a case of an adult female patient who discontinued mannose therapy in her adolescence. In adulthood she developed gastrointestinal problems, chronic anaemia and osteophytes in her knees.
磷酸甘露糖异构酶先天性糖基化障碍(MPI-CDG)由磷酸甘露糖异构酶缺乏引起。临床特征包括高胰岛素血症性低血糖、蛋白丢失性肠病、肝肿大和肝纤维化、消化症状以及凝血异常。该病采用补充甘露糖进行治疗。关于成人的长期预后情况描述不多。我们报告一例成年女性患者,她在青春期停用了甘露糖治疗。成年后,她出现了胃肠道问题、慢性贫血以及膝关节骨赘。