Barbeira Alvaro N, Melia Owen J, Liang Yanyu, Bonazzola Rodrigo, Wang Gao, Wheeler Heather E, Aguet François, Ardlie Kristin G, Wen Xiaoquan, Im Hae K
Section of Genetic Medicine, Department of Medicine, The University of Chicago, Chicago, Illinois.
Department of Human Genetics, The University of Chicago, Chicago, Illinois.
Genet Epidemiol. 2020 Sep 10;44(8):854-67. doi: 10.1002/gepi.22346.
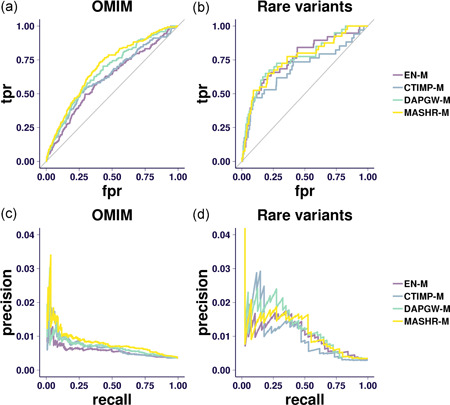
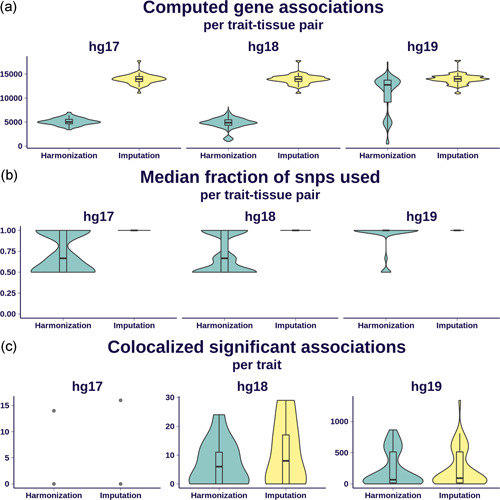
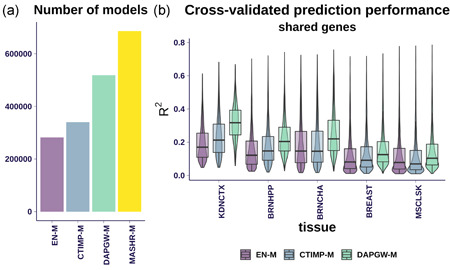
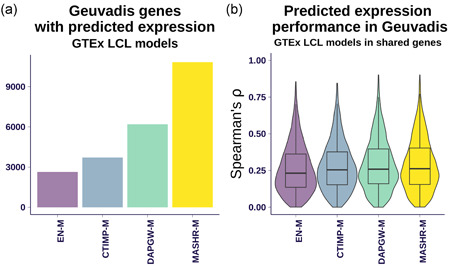
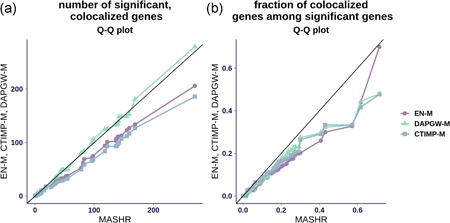
The integration of transcriptomic studies and genome-wide association studies (GWAS) via imputed expression has seen extensive application in recent years, enabling the functional characterization and causal gene prioritization of GWAS loci. However, the techniques for imputing transcriptomic traits from DNA variation remain underdeveloped. Furthermore, associations found when linking eQTL studies to complex traits through methods like PrediXcan can lead to false positives due to linkage disequilibrium between distinct causal variants. Therefore, the best prediction performance models may not necessarily lead to more reliable causal gene discovery. With the goal of improving discoveries without increasing false positives, we develop and compare multiple transcriptomic imputation approaches using the most recent GTEx release of expression and splicing data on 17,382 RNA-sequencing samples from 948 post-mortem donors in 54 tissues. We find that informing prediction models with posterior causal probability from fine-mapping (dap-g) and borrowing information across tissues (mashr) can lead to better performance in terms of number and proportion of significant associations that are colocalized and the proportion of silver standard genes identified as indicated by precision-recall and receiver operating characteristic curves. All prediction models are made publicly available at predictdb.org.
近年来,通过推测表达将转录组学研究与全基因组关联研究(GWAS)相结合得到了广泛应用,能够对GWAS位点进行功能表征并确定因果基因的优先级。然而,从DNA变异推测转录组特征的技术仍不发达。此外,当通过PrediXcan等方法将eQTL研究与复杂性状联系起来时,由于不同因果变异之间的连锁不平衡,发现的关联可能会导致假阳性。因此,最佳预测性能模型不一定能带来更可靠的因果基因发现。为了在不增加假阳性的情况下改进发现结果,我们使用来自54个组织中948名尸检供体的17382个RNA测序样本的最新GTEx表达和剪接数据版本,开发并比较了多种转录组推测方法。我们发现,利用精细定位(dap - g)的后验因果概率为预测模型提供信息并跨组织借用信息(mashr),在共定位的显著关联的数量和比例以及精确召回率和受试者工作特征曲线所表明的被鉴定为银标准基因的比例方面,可以带来更好的性能。所有预测模型均可在predictdb.org上公开获取。