Department of Psychiatry, Pamela Sklar Division of Psychiatric Genomics and Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY, 10029, USA.
Department of Genetics & Genomic Sciences and Institute for Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, 10029, NY, USA.
Nat Commun. 2019 Aug 23;10(1):3834. doi: 10.1038/s41467-019-11874-7.
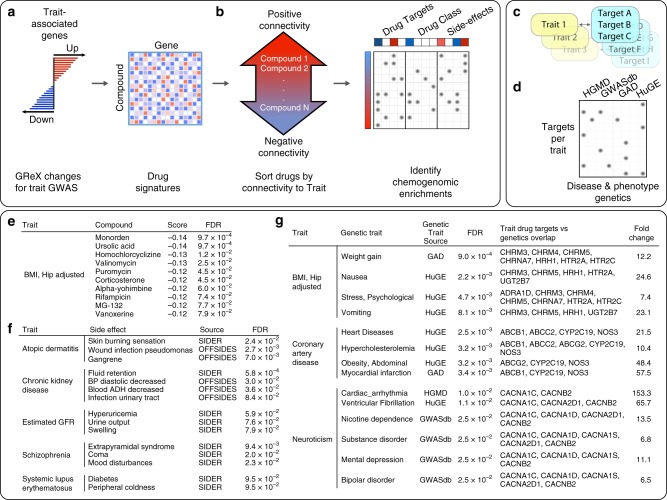
Transcriptome-wide association studies integrate gene expression data with common risk variation to identify gene-trait associations. By incorporating epigenome data to estimate the functional importance of genetic variation on gene expression, we generate a small but significant improvement in the accuracy of transcriptome prediction and increase the power to detect significant expression-trait associations. Joint analysis of 14 large-scale transcriptome datasets and 58 traits identify 13,724 significant expression-trait associations that converge on biological processes and relevant phenotypes in human and mouse phenotype databases. We perform drug repurposing analysis and identify compounds that mimic, or reverse, trait-specific changes. We identify genes that exhibit agonistic pleiotropy for genetically correlated traits that converge on shared biological pathways and elucidate distinct processes in disease etiopathogenesis. Overall, this comprehensive analysis provides insight into the specificity and convergence of gene expression on susceptibility to complex traits.
转录组关联研究将基因表达数据与常见风险变异相结合,以鉴定基因-性状关联。通过整合表观基因组数据来估计遗传变异对基因表达的功能重要性,我们可以在转录组预测的准确性上取得较小但显著的提高,并增加检测显著表达-性状关联的能力。对 14 个大规模转录组数据集和 58 个性状进行联合分析,确定了 13724 个显著的表达-性状关联,这些关联集中在人类和小鼠表型数据库中的生物学过程和相关表型上。我们进行药物重定位分析,并确定了模拟或逆转特定性状变化的化合物。我们鉴定出了表现出遗传相关性状的基因的激动性多效性,这些性状集中在共享的生物途径上,并阐明了疾病发病机制中的不同过程。总的来说,这项全面的分析为复杂性状易感性的基因表达的特异性和集中性提供了深入的了解。