Department of Pharmaceutical Chemistry, Cardiovascular Research Institute, University of California, San Francisco, San Francisco, CA, USA.
Department of Bioengineering and Therapeutic Sciences, University of California, San Francisco, San Francisco, CA, USA.
Nature. 2020 Oct;586(7827):145-150. doi: 10.1038/s41586-020-2761-3. Epub 2020 Sep 23.
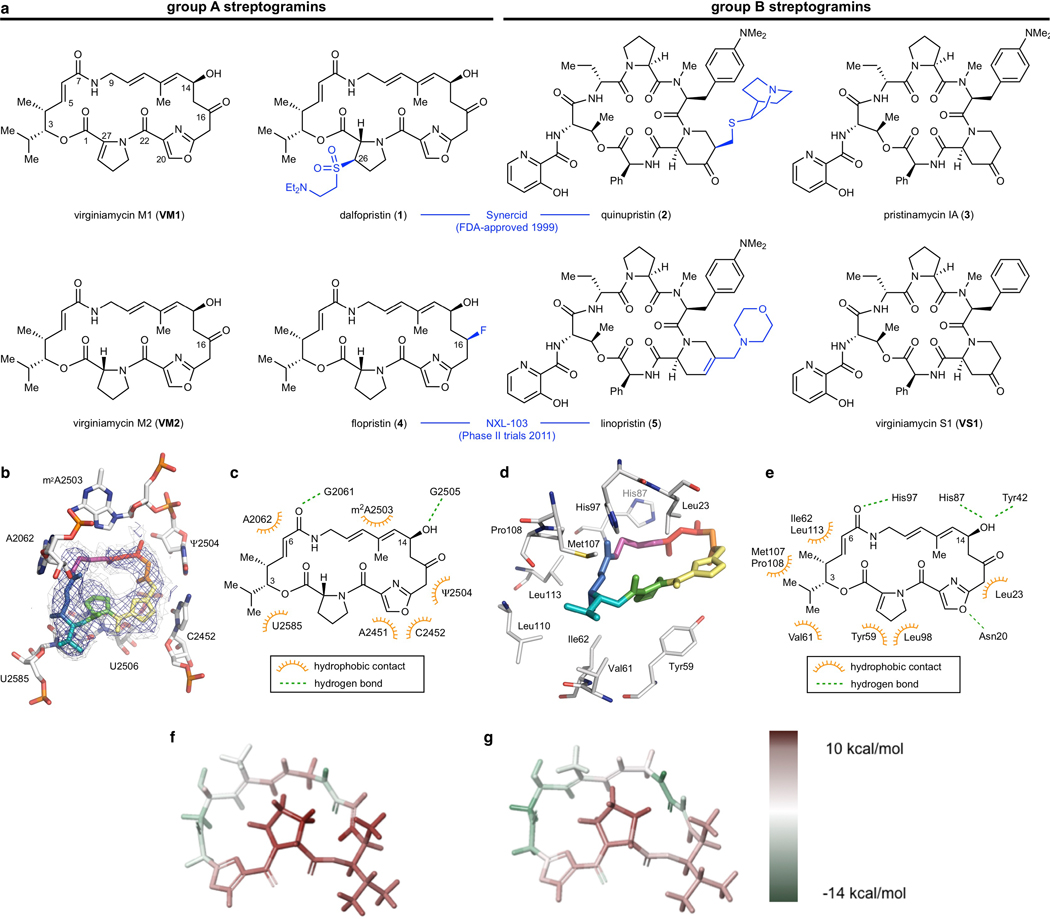
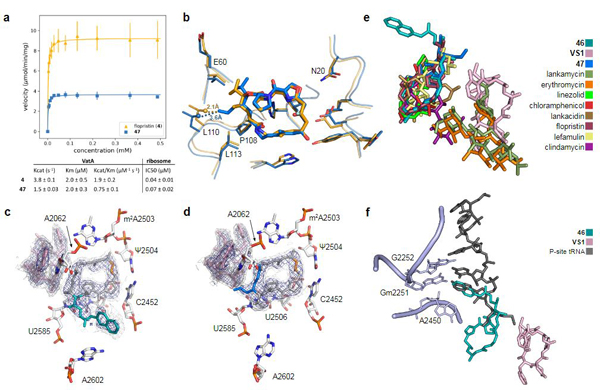
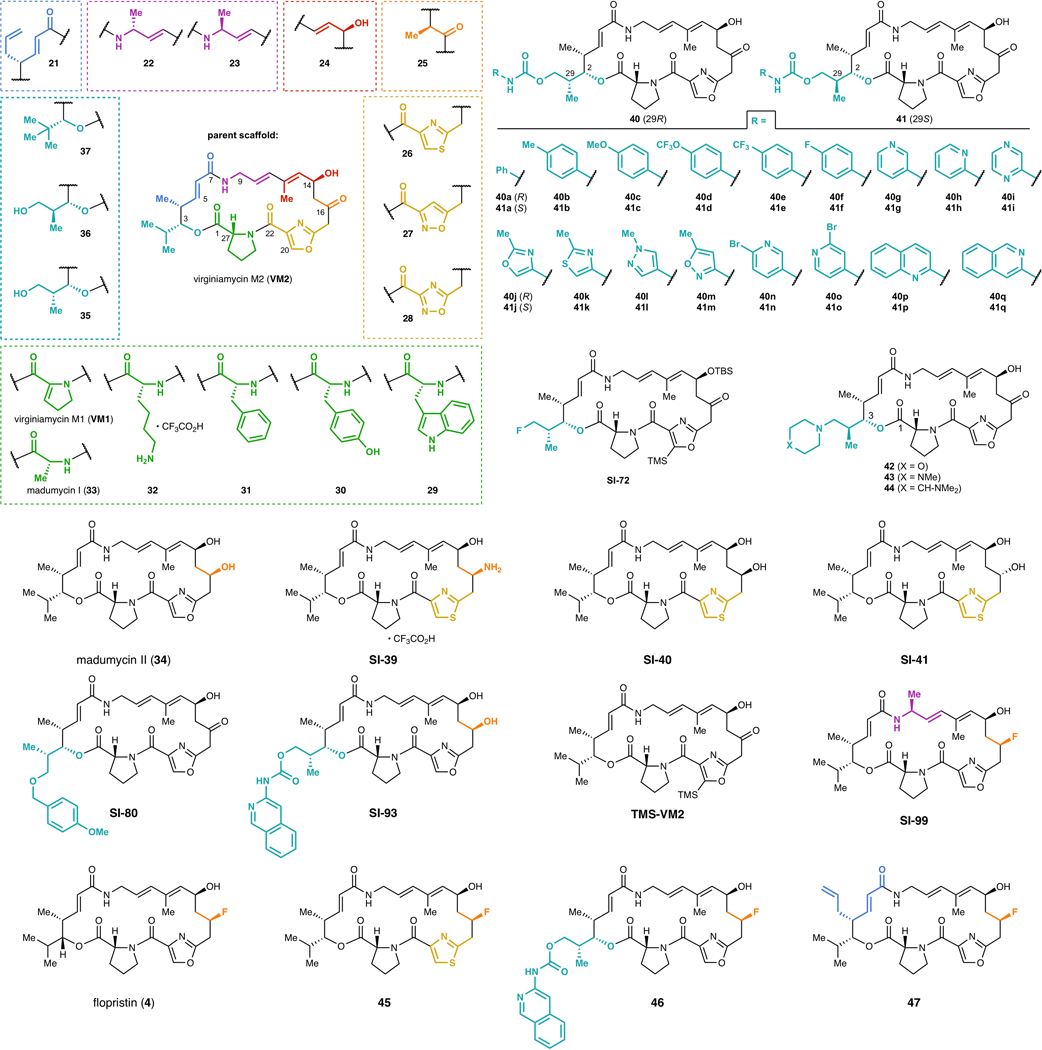
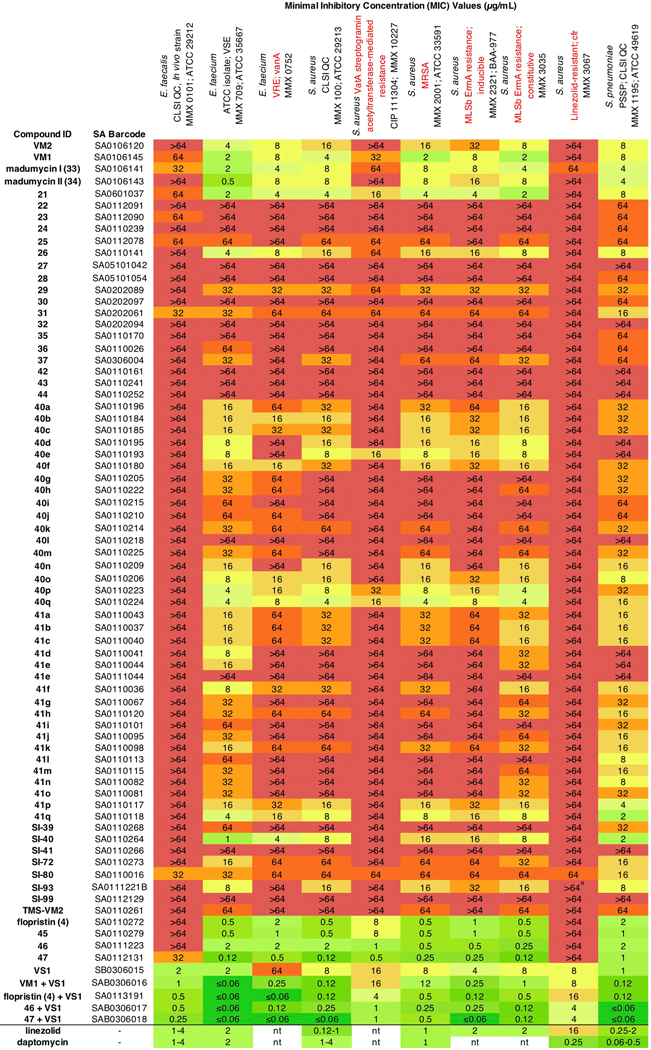
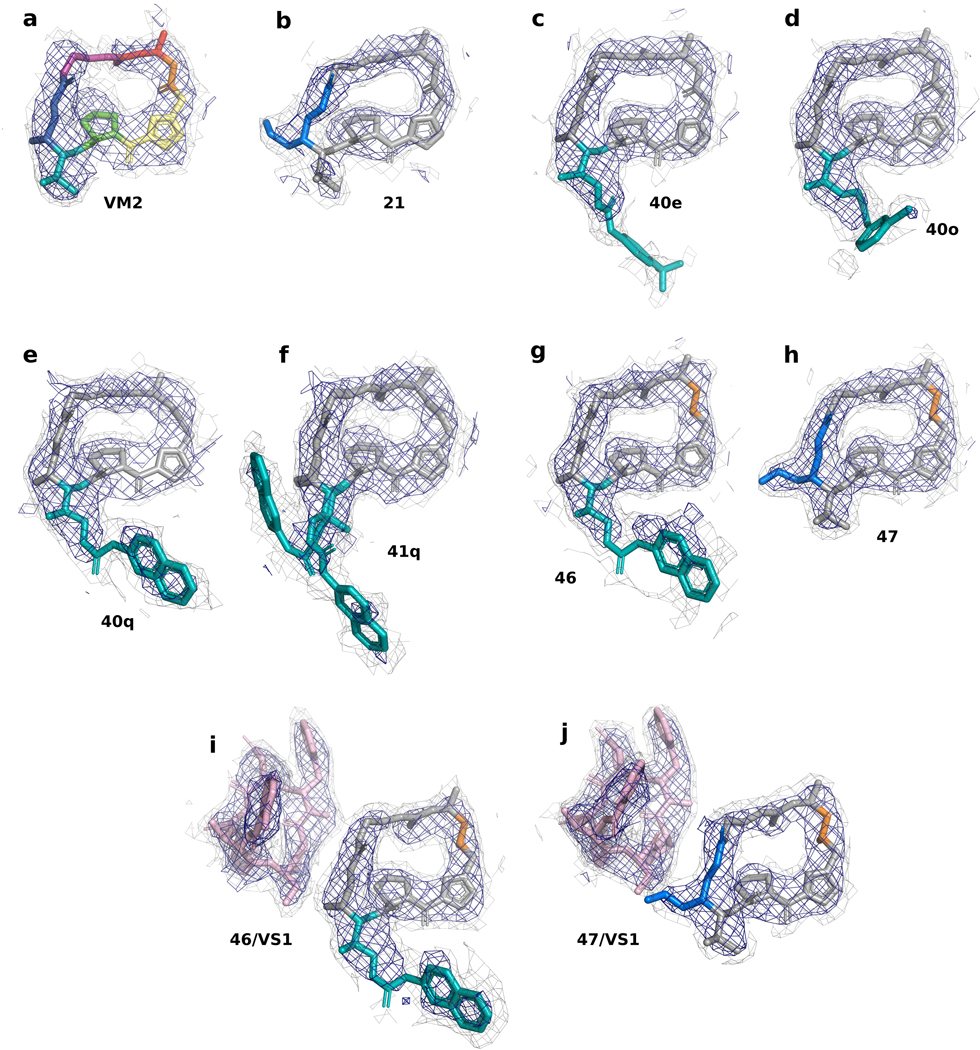
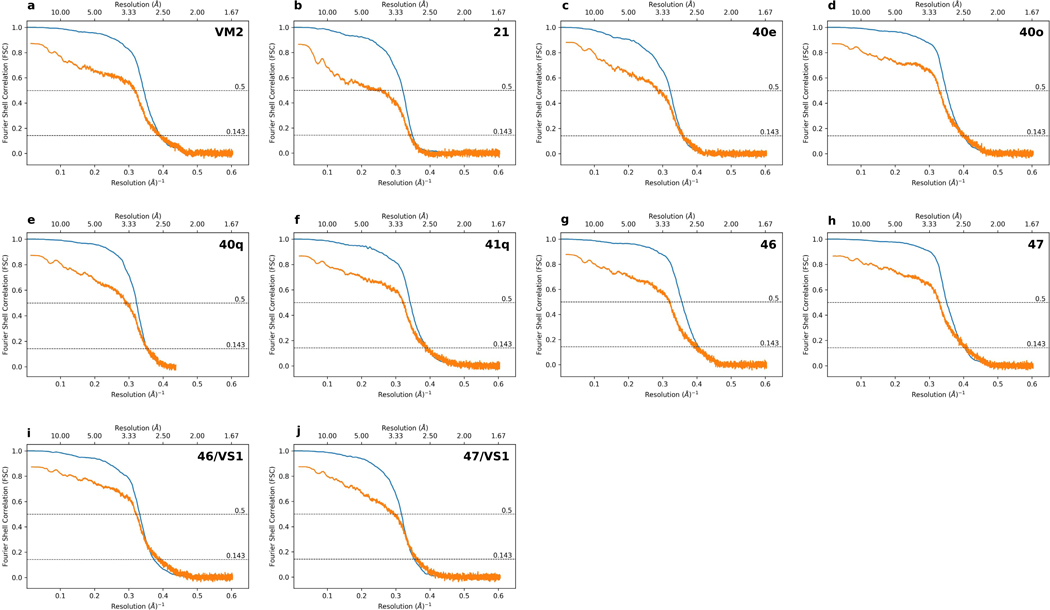
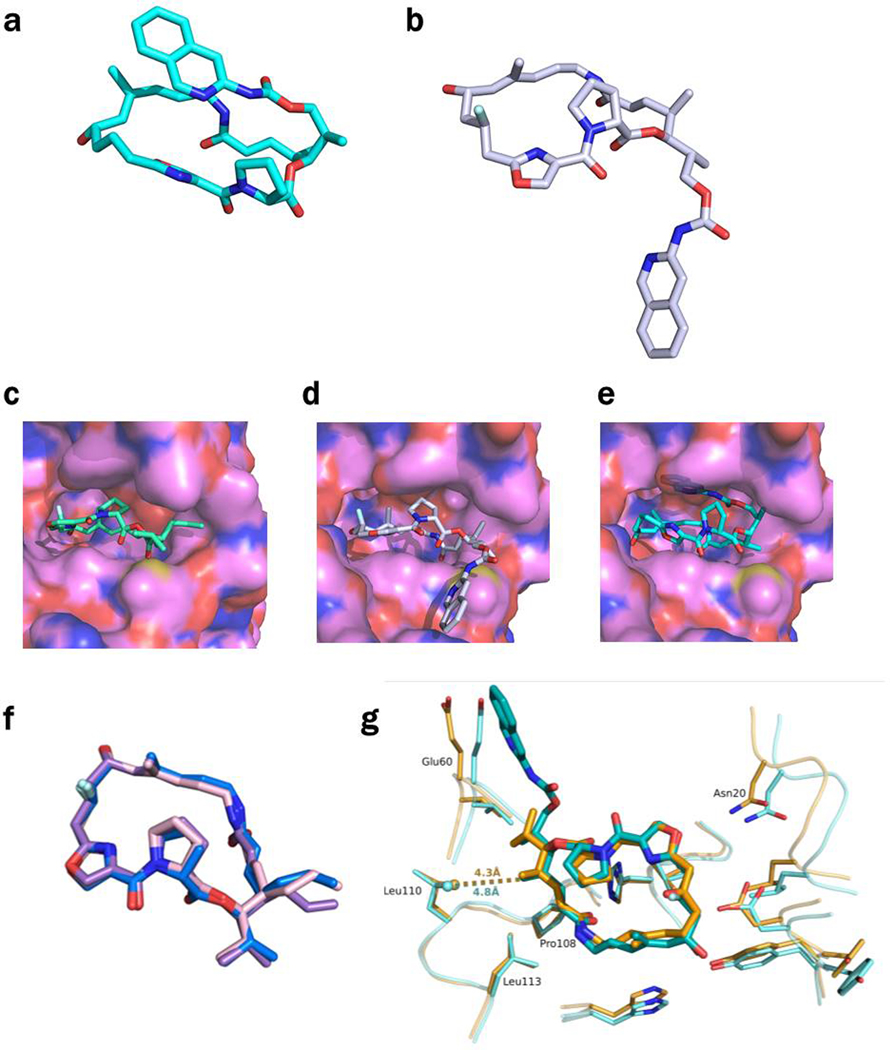
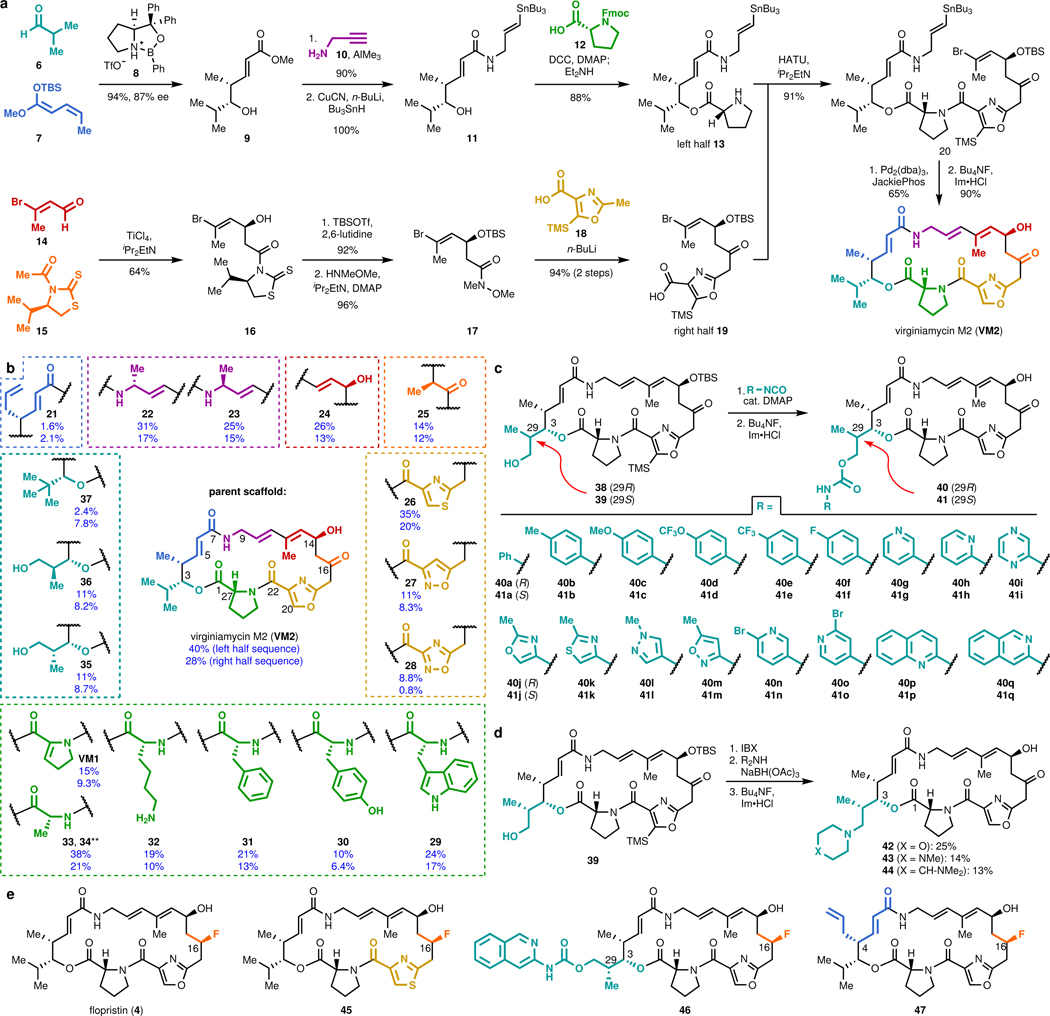
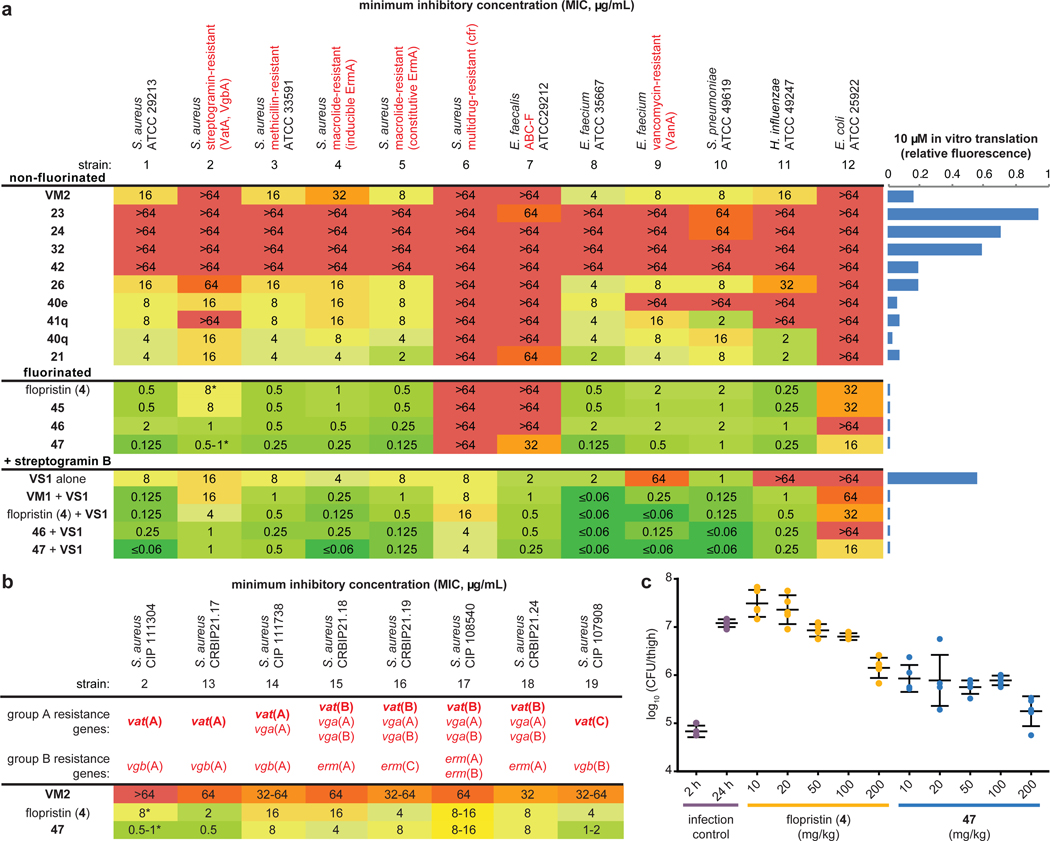
Natural products serve as chemical blueprints for most antibiotics in clinical use. The evolutionary process by which these molecules arise is inherently accompanied by the co-evolution of resistance mechanisms that shorten the clinical lifetime of any given class of antibiotics. Virginiamycin acetyltransferase (Vat) enzymes are resistance proteins that provide protection against streptogramins, potent antibiotics against Gram-positive bacteria that inhibit the bacterial ribosome. Owing to the challenge of selectively modifying the chemically complex, 23-membered macrocyclic scaffold of group A streptogramins, analogues that overcome the resistance conferred by Vat enzymes have not been previously developed. Here we report the design, synthesis, and antibacterial evaluation of group A streptogramin antibiotics with extensive structural variability. Using cryo-electron microscopy and forcefield-based refinement, we characterize the binding of eight analogues to the bacterial ribosome at high resolution, revealing binding interactions that extend into the peptidyl tRNA-binding site and towards synergistic binders that occupy the nascent peptide exit tunnel. One of these analogues has excellent activity against several streptogramin-resistant strains of Staphylococcus aureus, exhibits decreased rates of acetylation in vitro, and is effective at lowering bacterial load in a mouse model of infection. Our results demonstrate that the combination of rational design and modular chemical synthesis can revitalize classes of antibiotics that are limited by naturally arising resistance mechanisms.
天然产物为大多数临床使用的抗生素提供了化学蓝图。这些分子产生的进化过程,不可避免地伴随着耐药机制的共同进化,从而缩短了任何给定类别的抗生素的临床寿命。维吉尼亚霉素乙酰转移酶(Vat)酶是一种耐药蛋白,可提供对链球菌素的保护,链球菌素是一种针对革兰氏阳性菌的强效抗生素,可抑制细菌核糖体。由于选择性修饰结构复杂的 23 元大环 A 型链球菌素的挑战,以前尚未开发出克服 Vat 酶赋予的耐药性的类似物。在这里,我们报告了具有广泛结构变异性的 A 组链球菌素抗生素的设计、合成和抗菌评估。我们使用低温电子显微镜和基于力场的精修,以高分辨率表征了八种类似物与细菌核糖体的结合,揭示了延伸到肽基 tRNA 结合位点的结合相互作用,以及与占据新生肽出口隧道的协同结合物的结合相互作用。这些类似物之一对几种耐链球菌素的金黄色葡萄球菌菌株具有极好的活性,在体外表现出较低的乙酰化率,并且在感染小鼠模型中有效降低细菌负荷。我们的结果表明,合理设计和模块化化学合成的结合可以使受天然耐药机制限制的抗生素类别重新焕发活力。