Mendes Fernanda Rodrigues, Sobral Karine Marques, Culler Hebert Fabricio, Couto Samuel Campanelli Freitas, Pereira Juliana, Rocha Vanderson, Martinez Gracia Aparecida, Lage Luís Alberto de Pádua Covas
Department of Hematology, Hemotherapy and Cell Therapy, Medicine School, Sao Paulo University (FMUSP).
Fundação Pró-Sangue - Hemocentro de São Paulo, São Paulo, Brazil.
Medicine (Baltimore). 2020 Sep 25;99(39):e22299. doi: 10.1097/MD.0000000000022299.
Hemophagocytic lymphohistiocytosis (HLH) is a condition characterized by a hyperinflammatory state and persistent macrophage activation, resulting in reactive phagocytosis of the hematopoietic elements. In children, it is usually a hereditary disorder, while in adults it is usually acquired secondary to viral infections, collagenoses, or tumors. Although accounting for 10% of hematologic malignancies, HLH is rarely associated with multiple myeloma (MM) and other plasmacytic dyscrasias.
A 64-year-old Brazilian man seeked medical care with a 3-month history of intermittent fever, weight loss, night sweats, and progressive anemic symptoms.
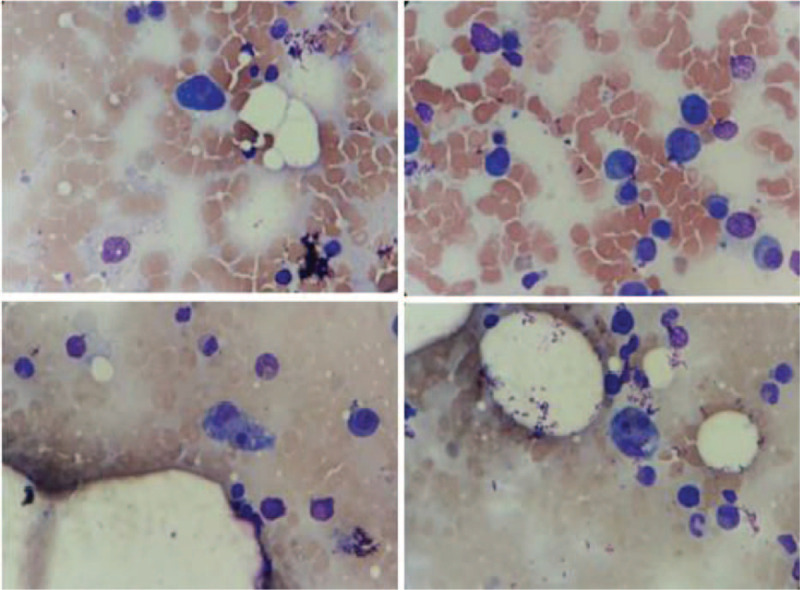
Total blood count showed severe bicytopenia (normocytic-normochromic anemia and thrombocytopenia), biochemical exams showed elevation of creatinine, as well as monoclonal peak in serum protein electrophoresis, high IgA dosage, and serum immunofixation with IgA kappa paraprotein. Bone marrow biopsy showed 30% of monoclonal and phenotypically anomalous plasmocytes, confirming the diagnosis of MM. Diagnosis of HLH was established by the presence of clinical and laboratory criteria: fever, splenomegaly, cytopenias, hypofibrinogenemia, hyperferritinemia, elevation of triglycerides, and several figures of erythrophagocytosis in bone marrow aspirate.
The patient experienced pulse therapy with methylprednisolone for hemophagocytic lymphohistiocytosis, followed by initial therapy for multiple myeloma with cyclophosphamide and dexamethasone.
Once the diagnosis of MM and secondary hemophagocytic syndrome was established, the patient had a rapid clinical deterioration despite the established therapeutic measures, evolving with cardiovascular failure, acute liver failure, acute disseminated intravascular coagulation, worsening renal dysfunction requiring dialysis support, respiratory dysfunction, and lowering of consciousness, characterizing rapid multiple organ dysfunction, ultimately leading to the death of the patient.
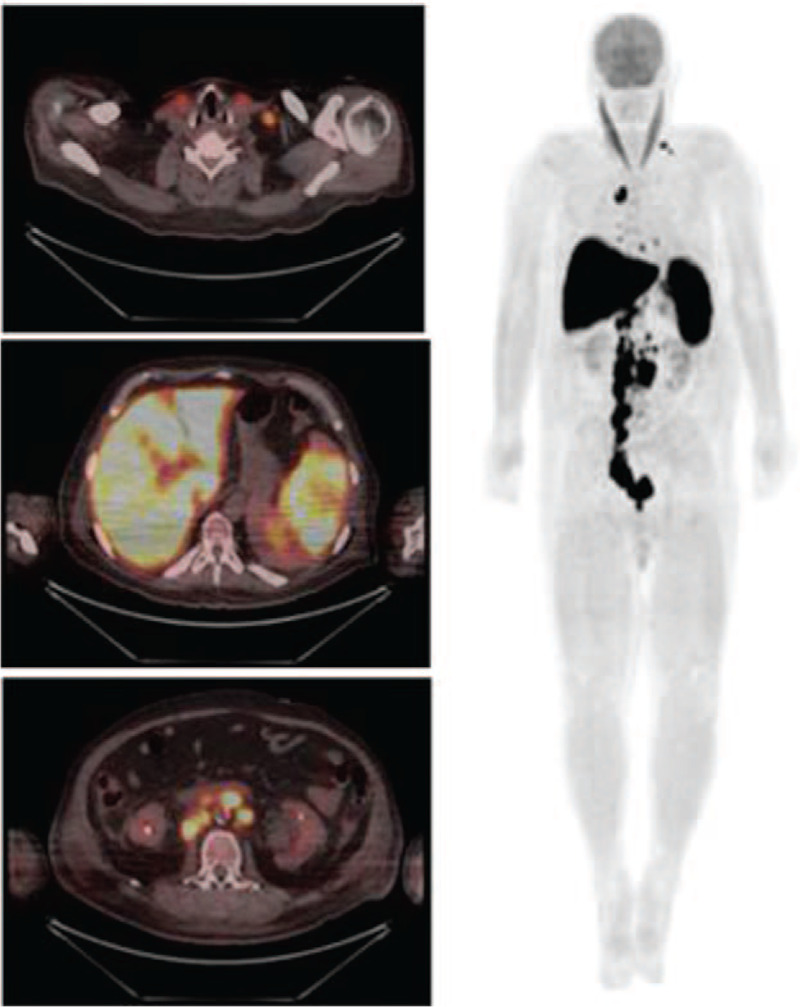
Here, we aimed to describe the sixth reported case of HLH associated with MM, according to cases cataloged in the PubMed database, and the first case evaluated by 18-fluordeoxyglucose positron emission tomography (18-FDG-PETCT).
Our case report seeks to provide support for a better clinical and laboratory characterization of this rare paraneoplastic entity associated with MM, and aims to call the attention of hematologists and intensivists to this condition that falls within the scope of the differential diagnosis of rapid onset multiple organ failure in patients with plasmacytic neoplasms.
噬血细胞性淋巴组织细胞增生症(HLH)是一种以高炎症状态和持续性巨噬细胞激活为特征的疾病,导致造血成分的反应性吞噬。在儿童中,它通常是一种遗传性疾病,而在成人中,它通常继发于病毒感染、胶原病或肿瘤。尽管HLH占血液系统恶性肿瘤的10%,但很少与多发性骨髓瘤(MM)和其他浆细胞异常增生相关。
一名64岁的巴西男子因间歇性发热、体重减轻、盗汗和进行性贫血症状3个月而寻求医疗护理。
全血细胞计数显示严重双血细胞减少(正细胞正色素性贫血和血小板减少),生化检查显示肌酐升高,血清蛋白电泳出现单克隆峰,IgA剂量升高,血清免疫固定显示IgA κ副蛋白。骨髓活检显示30%的单克隆和表型异常浆细胞,确诊为MM。通过临床和实验室标准确诊HLH:发热、脾肿大、血细胞减少、纤维蛋白原血症、高铁蛋白血症、甘油三酯升高以及骨髓穿刺涂片中出现多个红细胞吞噬现象。
患者接受了甲基强的松龙脉冲治疗以治疗噬血细胞性淋巴组织细胞增生症,随后采用环磷酰胺和地塞米松进行多发性骨髓瘤的初始治疗。
一旦确诊MM和继发性噬血细胞综合征,尽管采取了既定的治疗措施,患者的临床状况仍迅速恶化,发展为心血管衰竭、急性肝衰竭、急性弥散性血管内凝血、肾功能恶化需要透析支持、呼吸功能障碍和意识下降,表现为快速的多器官功能障碍,最终导致患者死亡。
在此,根据PubMed数据库中编目的病例,我们旨在描述第六例与MM相关的HLH报告病例,以及首例通过18-氟脱氧葡萄糖正电子发射断层扫描(18-FDG-PETCT)评估的病例。
我们的病例报告旨在为更好地临床和实验室表征这种与MM相关的罕见副肿瘤实体提供支持,并旨在引起血液学家和重症监护医生对这种属于浆细胞肿瘤患者快速发作多器官衰竭鉴别诊断范围内疾病的关注。