Department of Biostatistics and Translational Medicine, Medical University of Lodz, 15 Mazowiecka St., Lodz, 92-215, Poland.
Institute of Applied Computer Science, Lodz University of Technology, 18/22 Stefanowskiego St., Lodz, 90-537, Poland.
BMC Bioinformatics. 2020 Sep 29;21(1):425. doi: 10.1186/s12859-020-03743-8.
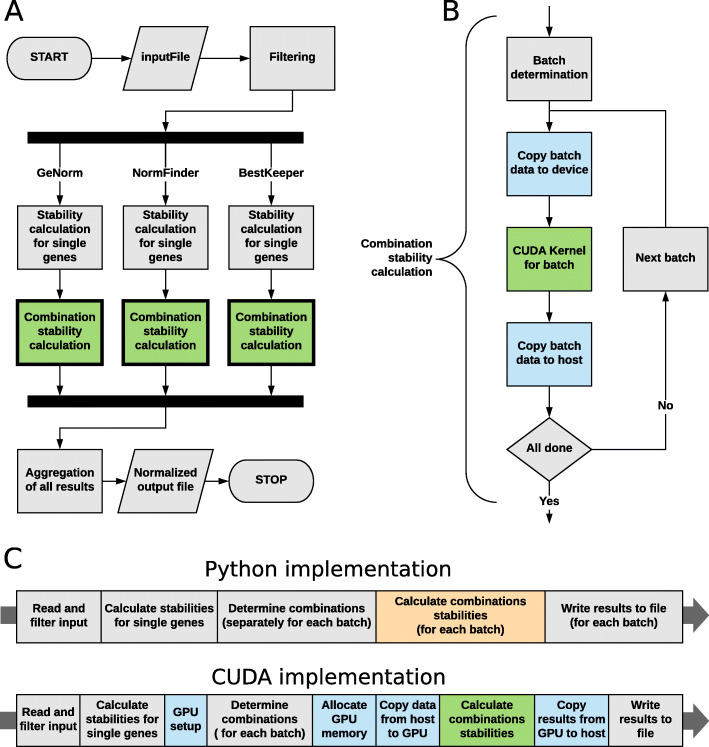
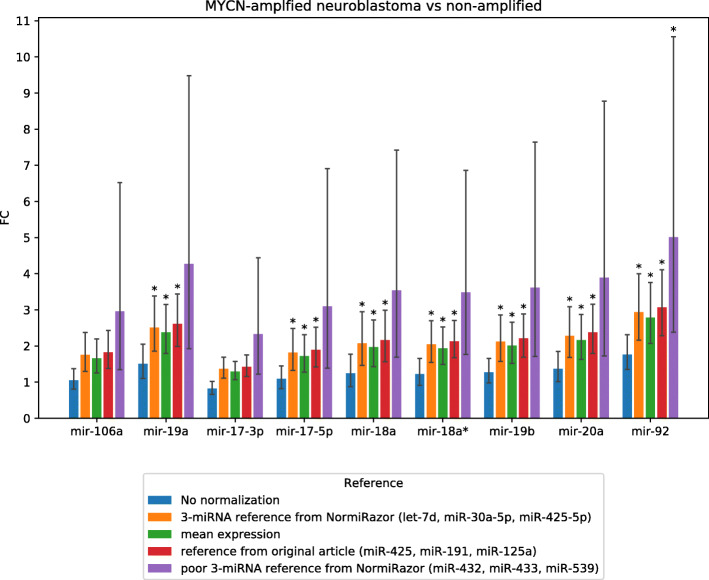
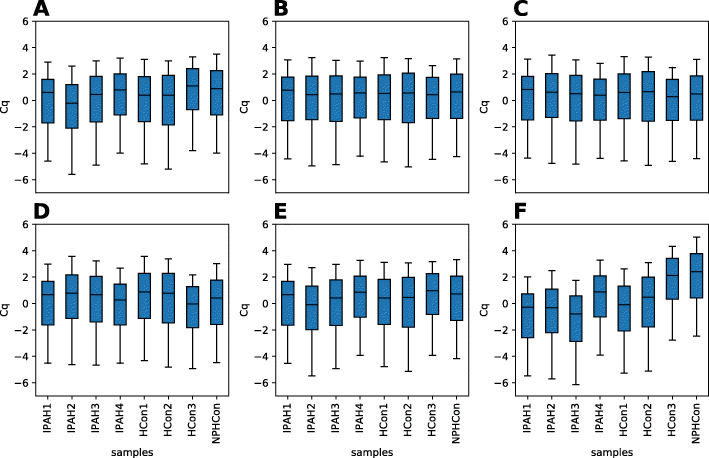
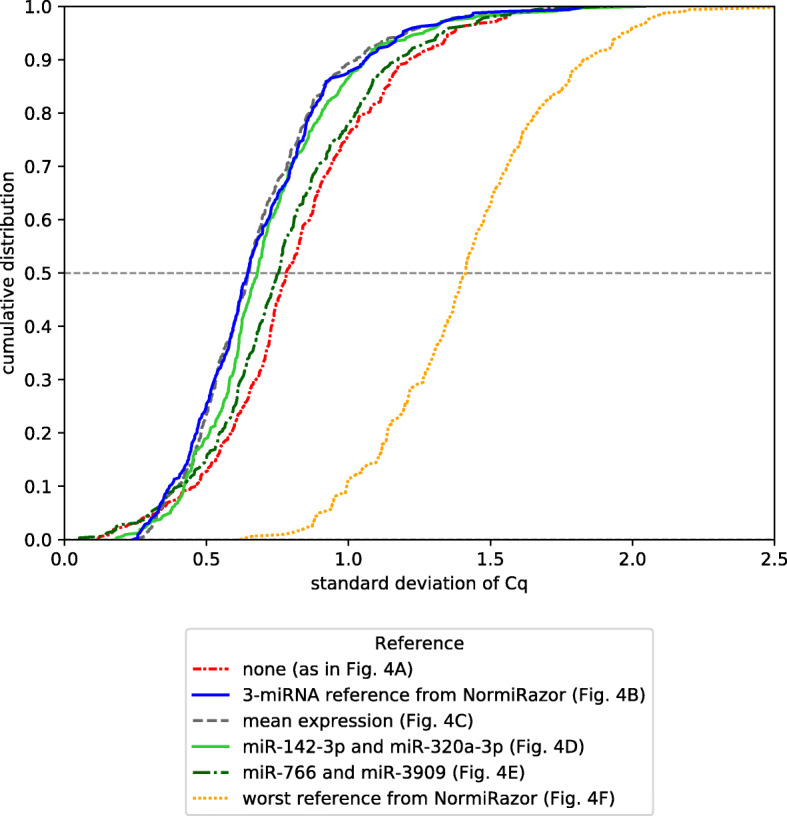
Multi-gene expression assays are an attractive tool in revealing complex regulatory mechanisms in living organisms. Normalization is an indispensable step of data analysis in all those studies, since it removes unwanted, non-biological variability from data. In targeted qPCR assays it is typically performed with respect to prespecified reference genes, but the lack of robust strategy of their selection is reported in literature, especially in studies concerning circulating microRNAs (miRNA). Unfortunately, this problem impedes translation of scientific discoveries on miRNA biomarkers into widely available laboratory assays. Previous studies concluded that averaged expressions of multi-miRNA combinations are more stable references than single genes. However, due to the number of such combinations the computational load is considerable and may be hindering for objective reference selection in large datasets. Existing implementations of normalization algorithms (geNorm, NormFinder and BestKeeper) have poor performance and may require days to compute stability values for all potential reference as the evaluation is performed sequentially.
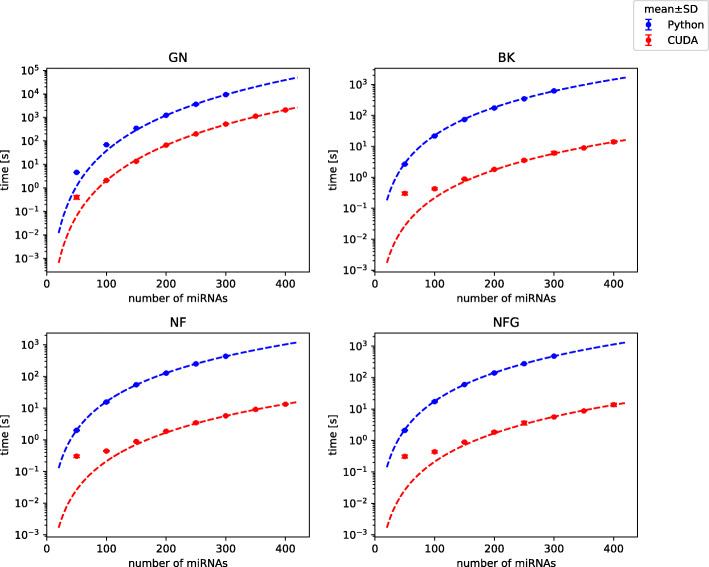
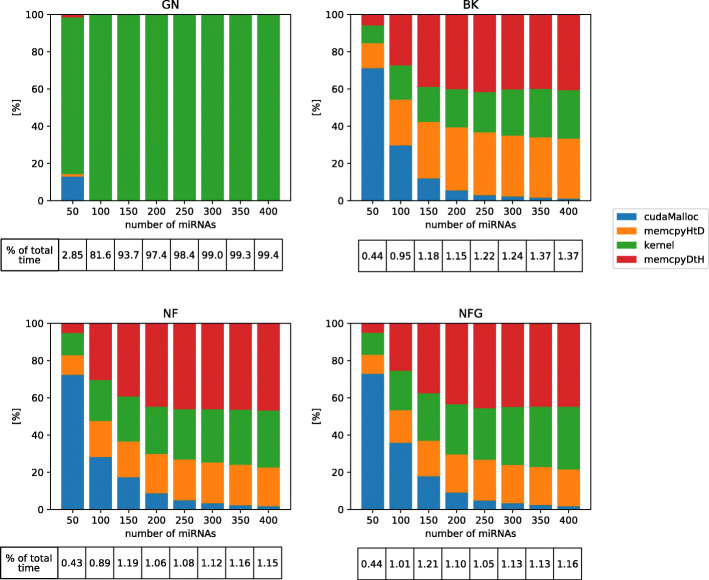
We designed NormiRazor - an integrative tool which implements those methods in a parallel manner on a graphics processing unit (GPU) using CUDA platform. We tested our approach on publicly available miRNA expression datasets. As a result, the times of executions on 8 datasets containing from 50 to 400 miRNAs (subsets of GSE68314) decreased 18.7 ±0.6 (mean ±SD), 104.7 ±4.2 and 76.5 ±2.2 times for geNorm, BestKeeper and NormFinder with respect to previous Python implementation. To allow for easy access to normalization pipeline for biomedical researchers we implemented NormiRazor as an online platform where a user could normalize their datasets based on the automatically selected references. It is available at norm.btm.umed.pl, together with instruction manual and exemplary datasets.
NormiRazor allows for an easy, informed choice of reference genes for qPCR transcriptomic studies. As such it can improve comparability and repeatability of experiments and in longer perspective help translate newly discovered biomarkers into readily available assays.
多基因表达分析是揭示生物体内复杂调控机制的一种有吸引力的工具。在所有这些研究中,标准化是数据分析不可或缺的一步,因为它可以从数据中去除不需要的、非生物学的变异性。在靶向 qPCR 分析中,通常是针对预定的参考基因进行的,但文献中报道了缺乏稳健的参考基因选择策略,尤其是在涉及循环 microRNA(miRNA)的研究中。不幸的是,这个问题阻碍了将 miRNA 生物标志物的科学发现转化为广泛可用的实验室检测。以前的研究得出结论,多 miRNA 组合的平均表达比单个基因更稳定。然而,由于组合数量众多,计算负荷相当大,可能会阻碍在大型数据集的客观参考选择。现有的归一化算法(geNorm、NormFinder 和 BestKeeper)的实现性能较差,可能需要数天时间才能为所有潜在的参考计算稳定性值,因为评估是顺序进行的。
我们设计了 NormiRazor-一种集成工具,它使用 CUDA 平台在图形处理单元(GPU)上以并行方式实现这些方法。我们在公开可用的 miRNA 表达数据集中测试了我们的方法。结果,在包含 50 到 400 个 miRNA(GSE68314 的子集)的 8 个数据集上的执行时间分别减少了 18.7±0.6(平均值±标准差)、104.7±4.2 和 76.5±2.2 倍,对于 geNorm、BestKeeper 和 NormFinder 分别相对于以前的 Python 实现。为了方便生物医学研究人员访问归一化管道,我们将 NormiRazor 实现为一个在线平台,用户可以在该平台上根据自动选择的参考进行数据集的归一化。它可在 norm.btm.umed.pl 上获得,同时提供使用手册和示例数据集。
NormiRazor 允许在 qPCR 转录组学研究中轻松、明智地选择参考基因。因此,它可以提高实验的可比性和可重复性,并在更长的时间内帮助将新发现的生物标志物转化为易于获得的检测。