Faculty of Computer Science, Dalhousie University, 6050 University Avenue, Halifax, Nova Scotia, B3H 4R2, Canada.
Department of Molecular Biology and Biochemistry, Simon Fraser University, 8888 University Drive, Burnaby, BC V5A 1S6, Canada.
Microb Genom. 2020 Oct;6(10). doi: 10.1099/mgen.0.000436.
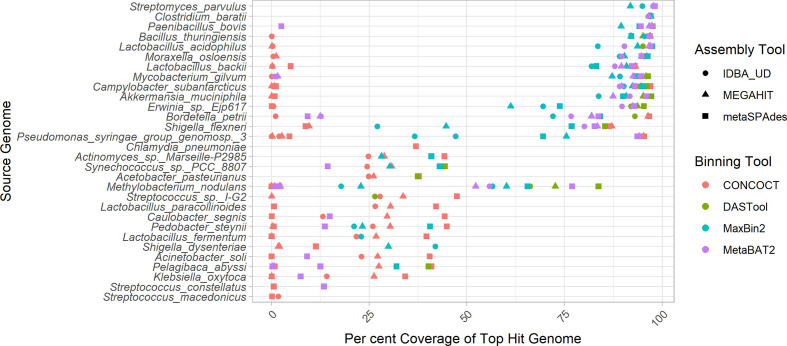
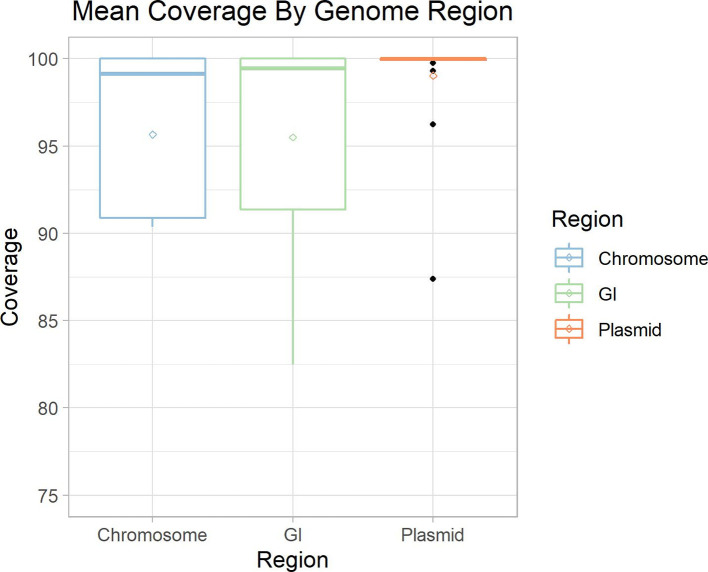
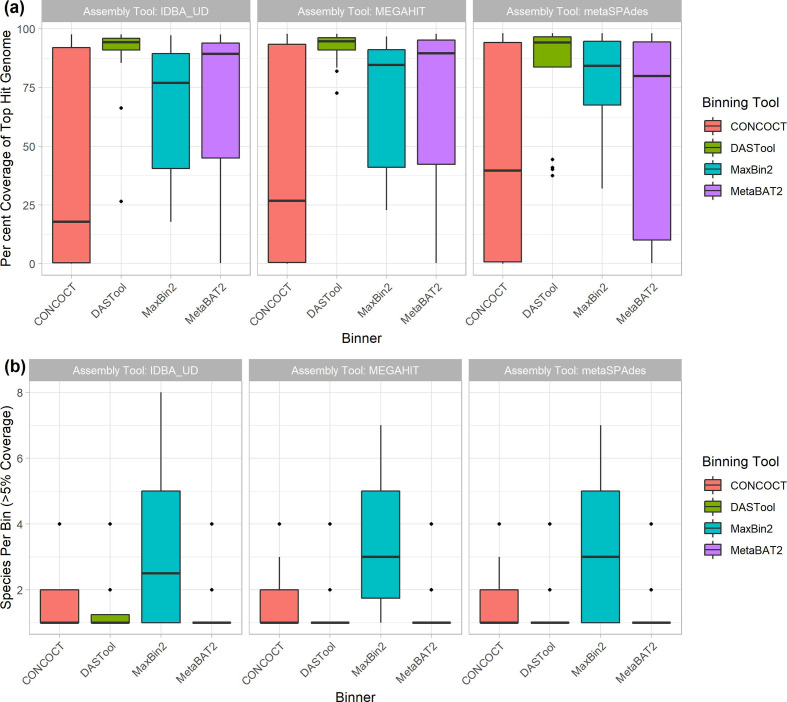
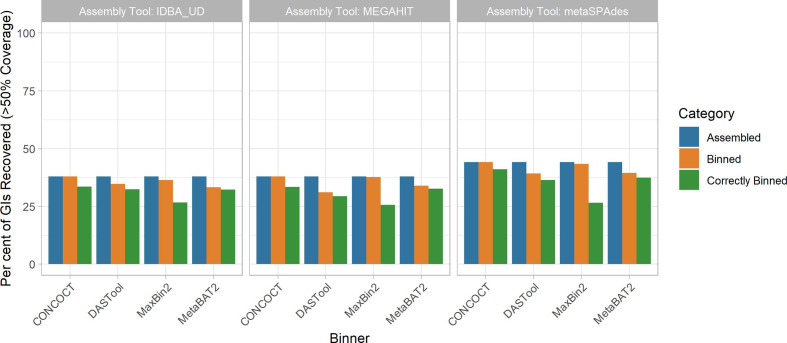
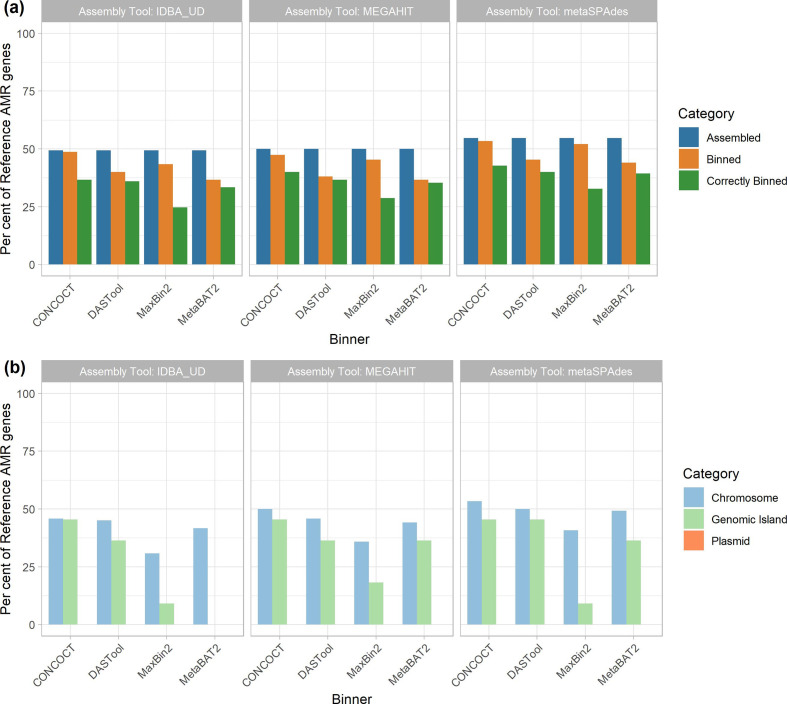
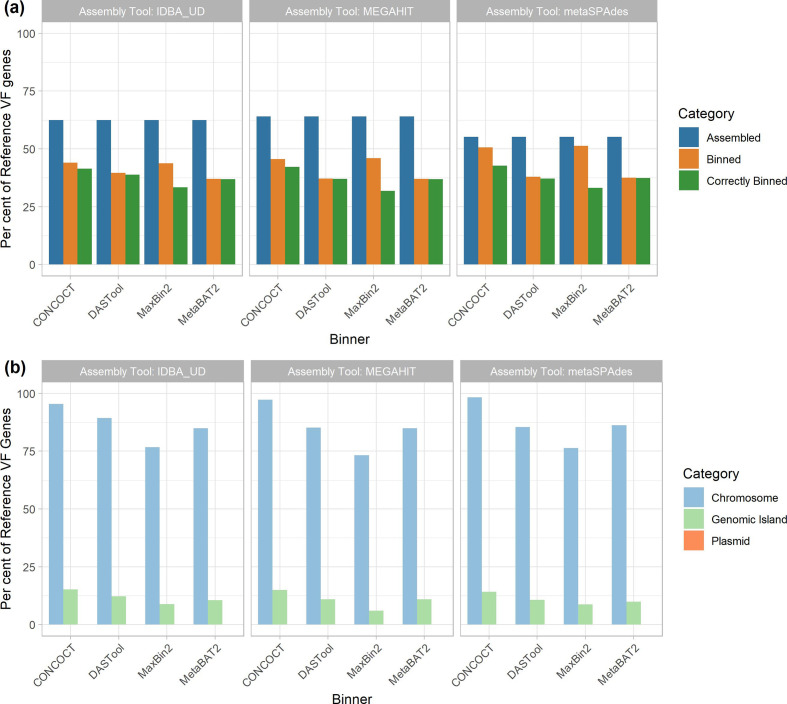
Metagenomic methods enable the simultaneous characterization of microbial communities without time-consuming and bias-inducing culturing. Metagenome-assembled genome (MAG) binning methods aim to reassemble individual genomes from this data. However, the recovery of mobile genetic elements (MGEs), such as plasmids and genomic islands (GIs), by binning has not been well characterized. Given the association of antimicrobial resistance (AMR) genes and virulence factor (VF) genes with MGEs, studying their transmission is a public-health priority. The variable copy number and sequence composition of MGEs makes them potentially problematic for MAG binning methods. To systematically investigate this issue, we simulated a low-complexity metagenome comprising 30 GI-rich and plasmid-containing bacterial genomes. MAGs were then recovered using 12 current prediction pipelines and evaluated. While 82-94 % of chromosomes could be correctly recovered and binned, only 38-44 % of GIs and 1-29 % of plasmid sequences were found. Strikingly, no plasmid-borne VF nor AMR genes were recovered, and only 0-45 % of AMR or VF genes within GIs. We conclude that short-read MAG approaches, without further optimization, are largely ineffective for the analysis of mobile genes, including those of public-health importance, such as AMR and VF genes. We propose that researchers should explore developing methods that optimize for this issue and consider also using unassembled short reads and/or long-read approaches to more fully characterize metagenomic data.
宏基因组方法能够在无需耗时且具有诱导偏差的培养的情况下同时对微生物群落进行特征分析。宏基因组组装基因组(MAG)分箱方法旨在从这些数据中重新组装单个基因组。然而,通过分箱来回收移动遗传元件(MGE),例如质粒和基因组岛(GI),尚未得到很好的描述。鉴于抗生素耐药性(AMR)基因和毒力因子(VF)基因与 MGE 相关,研究它们的传播是公共卫生的重点。MGE 的可变拷贝数和序列组成使其成为 MAG 分箱方法的潜在问题。为了系统地研究这个问题,我们模拟了一个包含 30 个 GI 丰富和含质粒的细菌基因组的低复杂度宏基因组。然后使用 12 种当前的预测管道来恢复 MAG 并进行评估。虽然可以正确恢复和分箱 82-94%的染色体,但仅找到 38-44%的 GI 和 1-29%的质粒序列。引人注目的是,没有回收到质粒携带的 VF 或 AMR 基因,并且仅在 GI 内回收了 0-45%的 AMR 或 VF 基因。我们得出结论,没有进一步优化的短读 MAG 方法在分析移动基因方面,包括那些具有公共卫生重要性的基因,例如 AMR 和 VF 基因,效果不大。我们建议研究人员探索开发针对该问题的方法,并考虑使用未组装的短读和/或长读方法来更全面地描述宏基因组数据。