Yang Xiu-Fang, Liu Guo-Sheng, Yi Bing
Department of Pediatrics and Neonatology, Zhongshan Hospital Affiliated to Sun Yat-Sen University, Zhongshan, Guangdong 528400, P.R. China.
Department of Neonatology, The First Affiliated Hospital of Jinan University, Guangzhou, Guangdong 510630, P.R. China.
Exp Ther Med. 2020 Nov;20(5):118. doi: 10.3892/etm.2020.9246. Epub 2020 Sep 21.
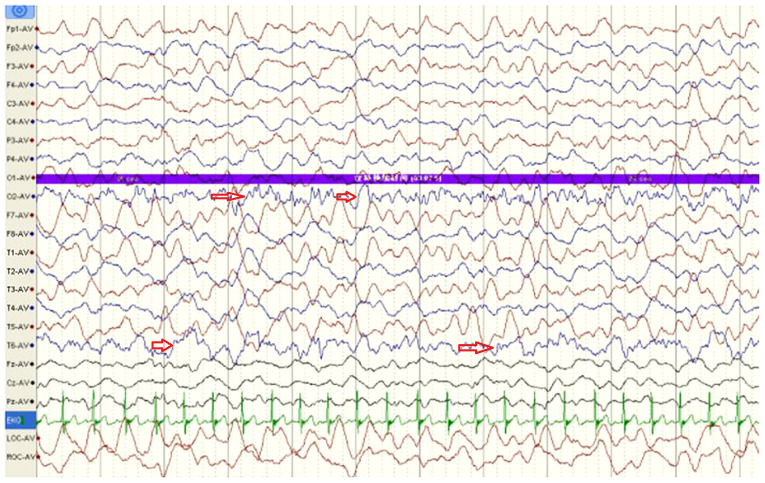
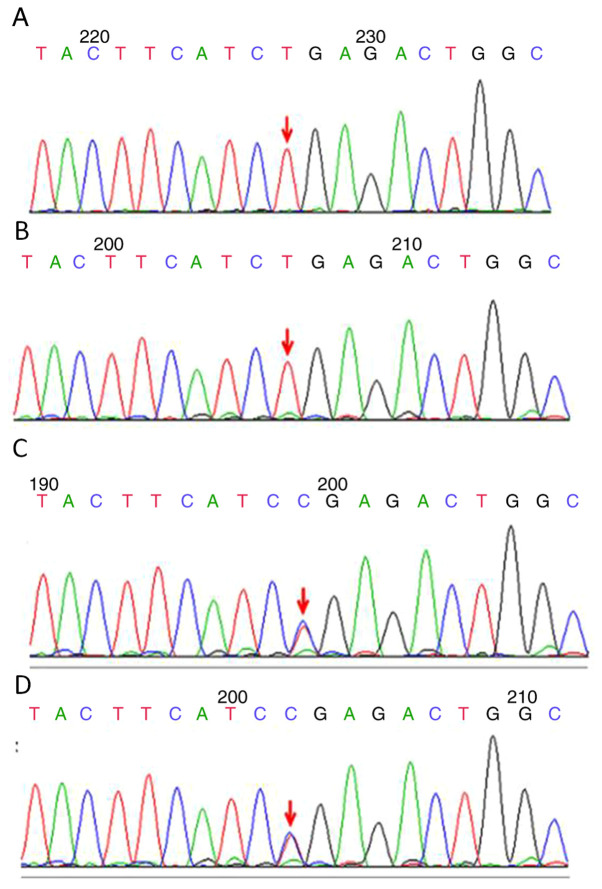
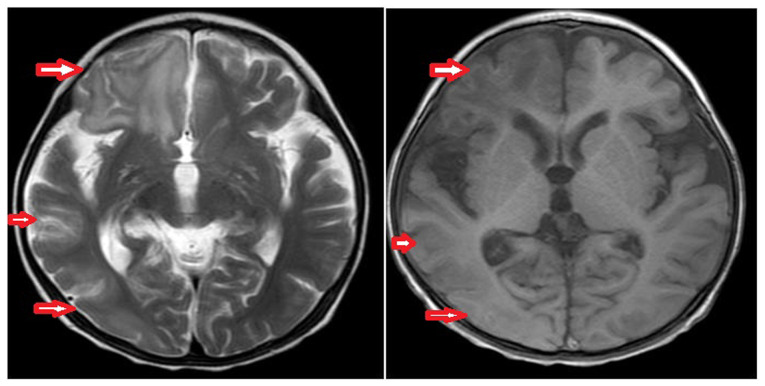
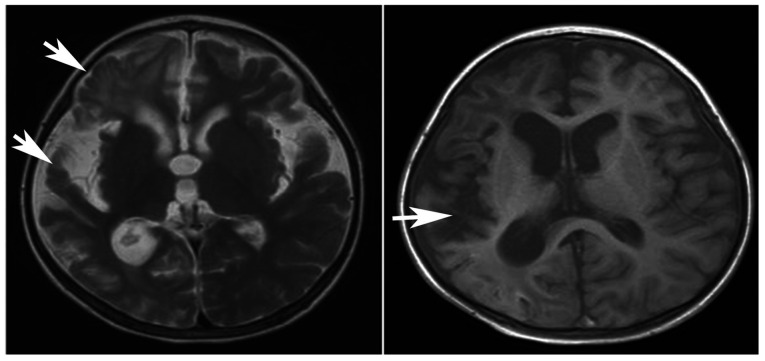
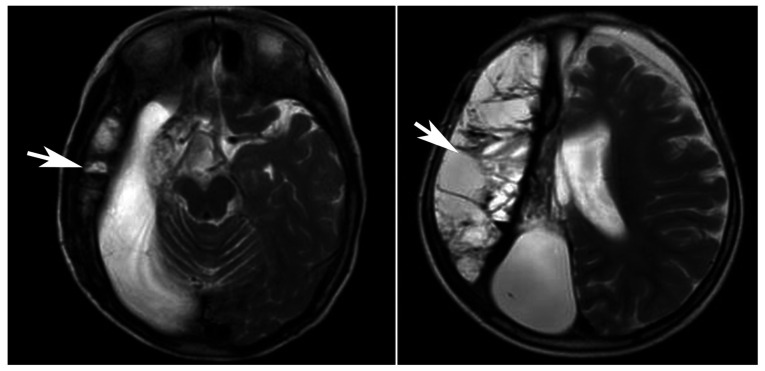
Primary carnitine deficiency (PCD) is a disorder of the carnitine cycle that results in defective fatty acid oxidation. When carnitine cannot be transported into the cells, fatty acid oxidation is impaired, resulting a variety of symptoms, such as chronic muscle weakness, cardiomyopathy, hypoglycemia and liver dysfunction. The clinical manifestations and outcomes of different cases with PCD vary among patients. The present case report focused on two sisters with PCD. The younger sister presented with intractable epilepsy, and the older sister presented with reversible metabolic cardiomyopathy. Potential mutations in the SLC22A5 gene were investigated within the family, and a nonsense mutation [c.760C>T (p.R254X)] was identified in four family members. The two sisters harbored homozygous mutations, whereas their parents presented heterozygous mutations. Metabolic disease screening revealed low plasma free carnitine levels (<5 µmol/l) in the two sisters. The plasma free carnitine levels of their parents were normal, and they were asymptomatic. PCD in the two patients was managed using oral levocarnitine. The metabolic cardiomyopathy of the older sister improved following 3 months of treatment. However, the epilepsy of the younger sister was recurrent with oral antiepileptic therapy lasting one year and eight months, and epilepsy was finally controlled following right cerebral resection. The present case report demonstrated that the clinical manifestations presented by patients with PCD within the same family were different. The results indicated that treatment with levocarnitine supplementation should be initiated as soon as possible before irreversible organ damage occurs. In addition, metabolic decompensation and cardiac muscle functions were improved following carnitine supplementation. The resection of the severely diseased unilateral brain combined with carnitine supplementation and antiepileptic therapy may be an effective treatment for PCD with intractable epilepsy complications.
原发性肉碱缺乏症(PCD)是一种肉碱循环障碍疾病,会导致脂肪酸氧化缺陷。当肉碱无法转运进入细胞时,脂肪酸氧化就会受损,从而引发多种症状,如慢性肌无力、心肌病、低血糖和肝功能障碍。不同PCD病例的临床表现和预后在患者之间存在差异。本病例报告聚焦于两名患有PCD的姐妹。妹妹表现为难治性癫痫,而姐姐表现为可逆性代谢性心肌病。对该家族进行了SLC22A5基因潜在突变的研究,在四名家庭成员中发现了一个无义突变[c.760C>T (p.R254X)]。两姐妹为纯合突变,而她们的父母为杂合突变。代谢疾病筛查显示,两姐妹的血浆游离肉碱水平较低(<5 μmol/l)。她们父母的血浆游离肉碱水平正常,且无症状。两名患者的PCD均采用口服左卡尼汀进行治疗。治疗3个月后,姐姐的代谢性心肌病有所改善。然而,妹妹的癫痫在口服抗癫痫治疗持续一年零八个月后仍反复发作,最终在右大脑切除术后癫痫得到控制。本病例报告表明,同一家庭中PCD患者的临床表现有所不同。结果表明,应在不可逆器官损伤发生之前尽快开始补充左卡尼汀进行治疗。此外,补充肉碱后代谢失代偿和心肌功能得到改善。切除严重病变的单侧大脑联合补充肉碱和抗癫痫治疗可能是治疗伴有难治性癫痫并发症的PCD的有效方法。