Kubota Tomoya, Wu Fenfen, Vicart Savine, Nakaza Maki, Sternberg Damien, Watanabe Daisuke, Furuta Mitsuru, Kokunai Yosuke, Abe Tatsuya, Kokubun Norito, Fontaine Bertrand, Cannon Stephen C, Takahashi Masanori P
Division of Health Sciences, Department of Functional Diagnostic Science, Osaka University Graduate School of Medicine, 1-7, Yamadaoka, Suita, Osaka, 5650871, Japan.
Department of Neurology, Osaka University Graduate School of Medicine, Suita, Osaka, Japan.
Brain Commun. 2020 Jul 16;2(2):fcaa103. doi: 10.1093/braincomms/fcaa103. eCollection 2020.
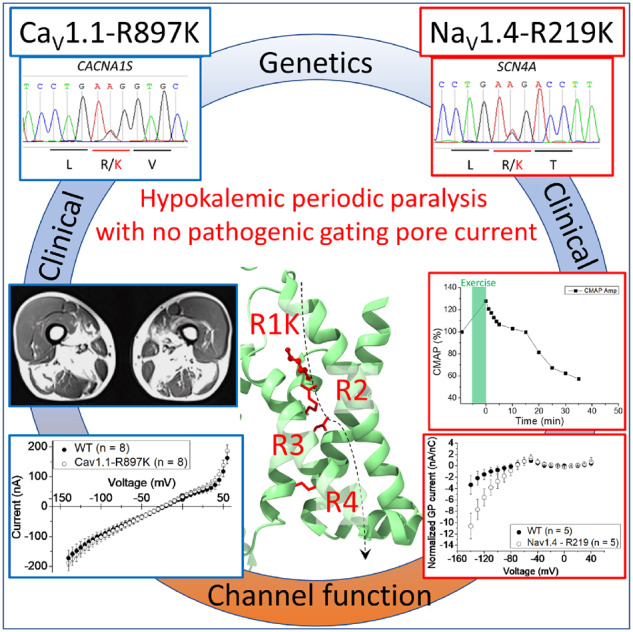
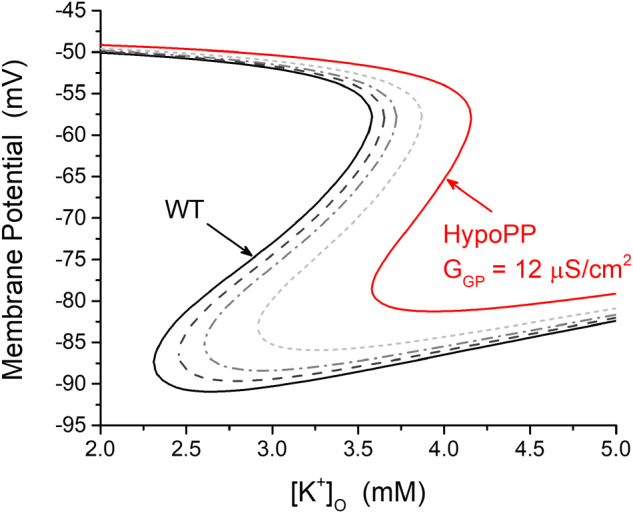
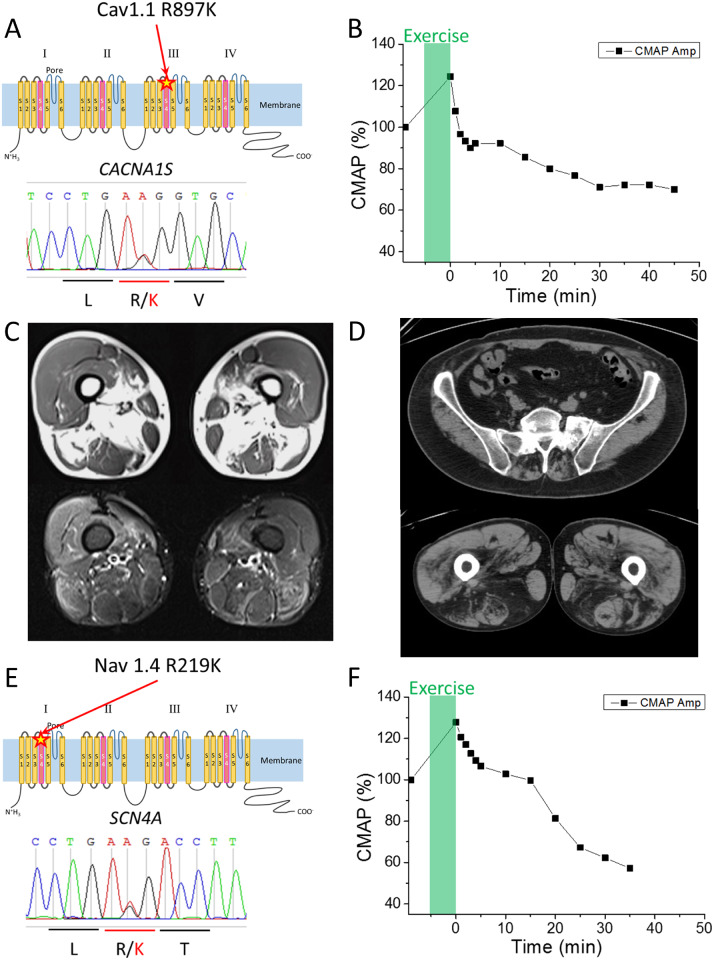
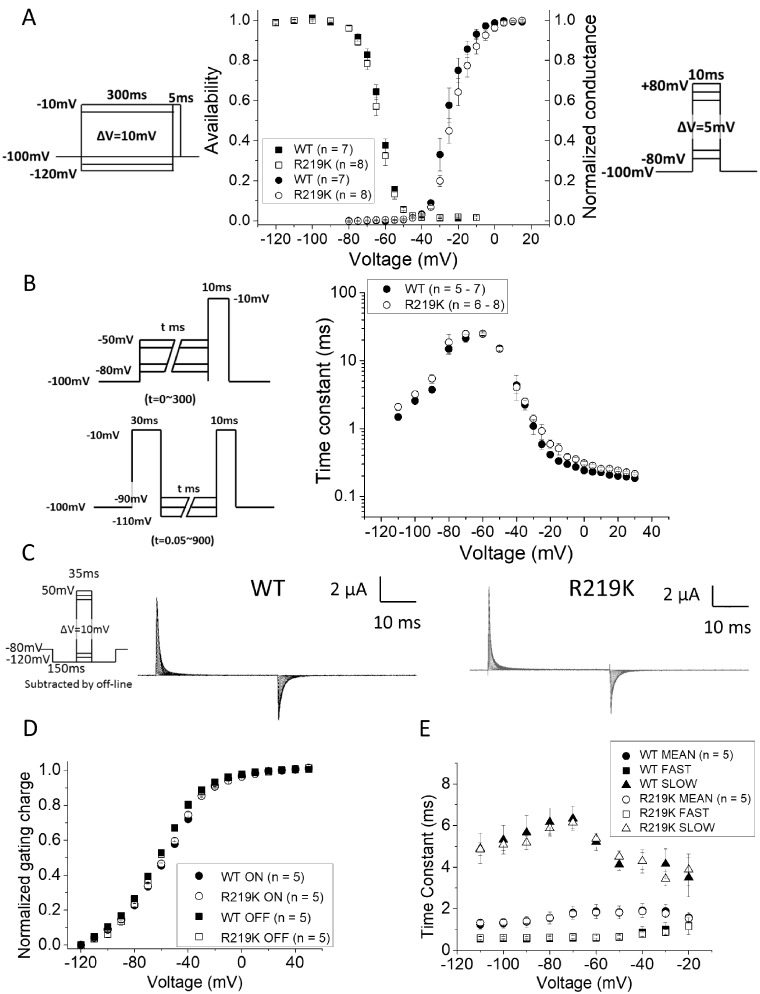
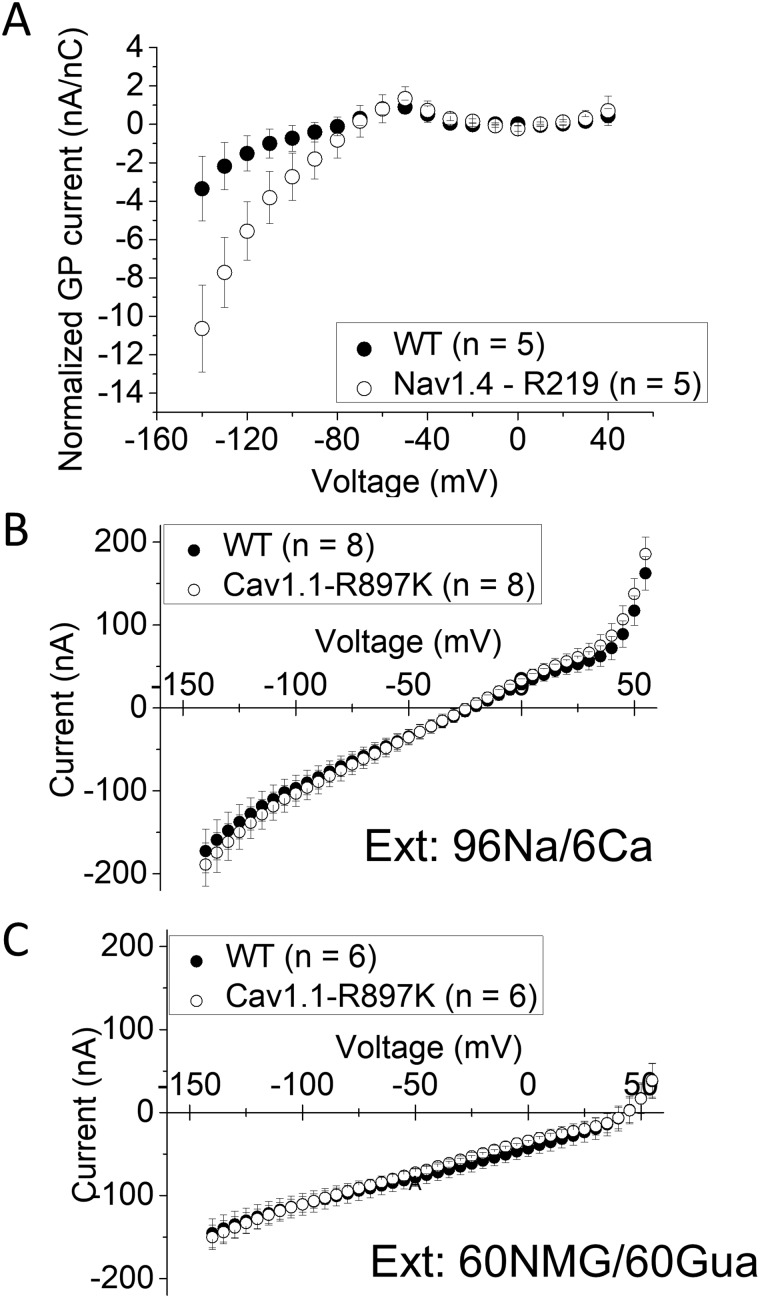
Familial hypokalaemic periodic paralysis is a rare skeletal muscle disease caused by the dysregulation of sarcolemmal excitability. Hypokalaemic periodic paralysis is characterized by repeated episodes of paralytic attacks with hypokalaemia, and several variants in coding for Ca1.1 and coding for Na1.4 have been established as causative mutations. Most of the mutations are substitutions to a non-charged residue, from the positively charged arginine (R) in transmembrane segment 4 (S4) of a voltage sensor in either Ca1.1 or Na1.4. Mutant channels have aberrant leak currents called 'gating pore currents', and the widely accepted consensus is that this current is the essential pathological mechanism that produces susceptibility to anomalous depolarization and failure of muscle excitability during a paralytic attack. Here, we have identified five hypokalaemic periodic paralysis cases from two different ethnic backgrounds, Japanese and French, with charge-preserving substitutions in S4 from arginine, R, to lysine, K. An R to K substitution has not previously been reported for any other hypokalaemic periodic paralysis families. One case is R219K in Na1.4, which is located at the first charge in S4 of Domain I. The other four cases all have R897K in Ca1.1, which is located at the first charge in S4 of Domain III. Gating pore currents were not detected in expression studies of Ca1.1-R897K. Na1.4-R219K mutant channels revealed a distinct, but small, gating pore current. Simulation studies indicated that the small-amplitude gating pore current conducted by Na1.4-R219K is not likely to be sufficient to be a risk factor for depolarization-induced paralytic attacks. Our rare cases with typical hypokalaemic periodic paralysis phenotypes do not fit the canonical view that the essential defect in hypokalaemic periodic paralysis mutant channels is the gating pore current and raise the possibility that hypokalaemic periodic paralysis pathogenesis might be heterogeneous and diverse.
家族性低钾性周期性麻痹是一种由肌膜兴奋性失调引起的罕见骨骼肌疾病。低钾性周期性麻痹的特征是伴有低钾血症的反复麻痹发作,并且已确定编码Ca1.1和编码Na1.4的几个变体为致病突变。大多数突变是从Ca1.1或Na1.4电压传感器跨膜段4(S4)中带正电荷的精氨酸(R)替换为不带电荷的残基。突变通道具有异常的泄漏电流,称为“门控孔电流”,并且广泛接受的共识是,这种电流是在麻痹发作期间产生对异常去极化的易感性和肌肉兴奋性丧失的基本病理机制。在这里,我们从两个不同的种族背景(日本人和法国人)中鉴定出五例低钾性周期性麻痹病例,其S4中存在从精氨酸(R)到赖氨酸(K)的电荷保留替换。以前尚未报道过任何其他低钾性周期性麻痹家族有R到K的替换。一例是Na1.4中的R219K,位于结构域I的S4中的第一个电荷处。其他四例均在Ca1.1中有R897K,位于结构域III的S4中的第一个电荷处。在Ca1.1-R897K的表达研究中未检测到门控孔电流。Na1.4-R219K突变通道显示出明显但较小的门控孔电流。模拟研究表明,由Na1.4-R219K传导的小幅度门控孔电流不太可能足以成为去极化诱导的麻痹发作的危险因素。我们具有典型低钾性周期性麻痹表型的罕见病例不符合低钾性周期性麻痹突变通道的基本缺陷是门控孔电流的传统观点,并提出低钾性周期性麻痹发病机制可能是异质性和多样性的可能性。