Fouka Maria, Mavroeidi Panagiota, Tsaka Grigoria, Xilouri Maria
Center of Clinical Research, Experimental Surgery and Translational Research, Biomedical Research Foundation of the Academy of Athens, Athens, Greece.
Front Cell Dev Biol. 2020 Sep 4;8:559791. doi: 10.3389/fcell.2020.559791. eCollection 2020.
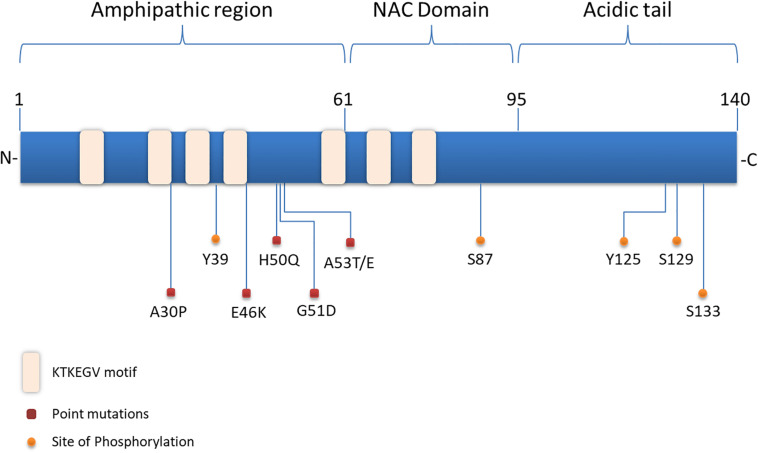
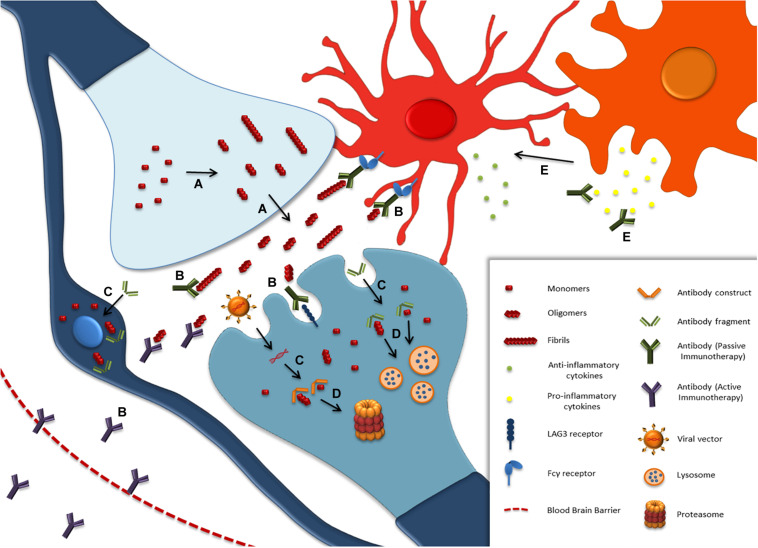
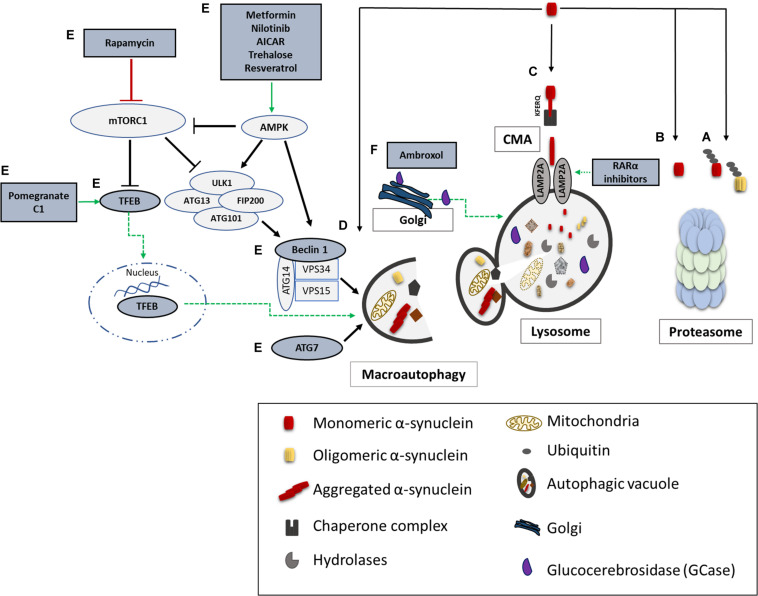
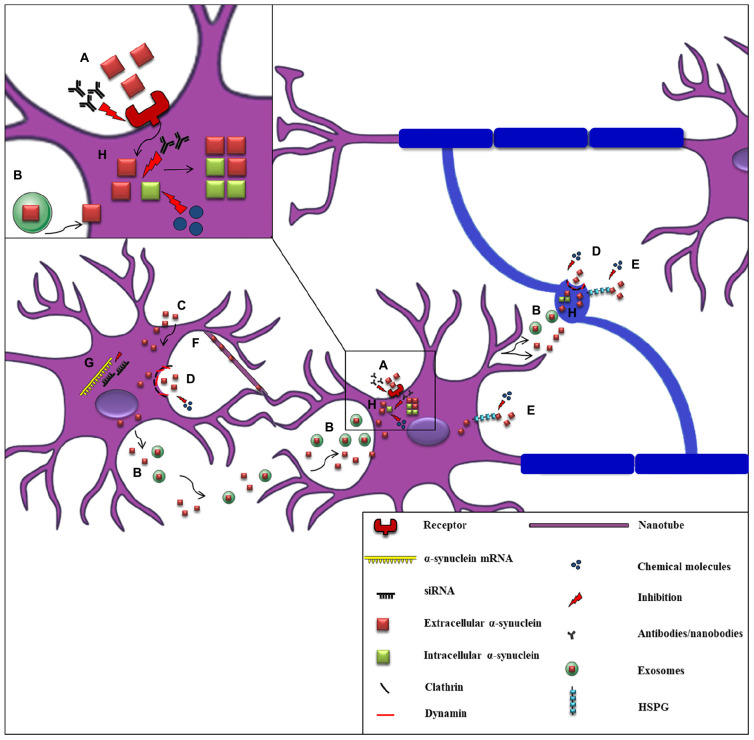
Parkinson's disease (PD), multiple system atrophy (MSA) and Dementia with Lewy bodies (DLB) represent pathologically similar, progressive neurodegenerative disorders characterized by the pathological aggregation of the neuronal protein α-synuclein. PD and DLB are characterized by the abnormal accumulation and aggregation of α-synuclein in proteinaceous inclusions within neurons named Lewy bodies (LBs) and Lewy neurites (LNs), whereas in MSA α-synuclein inclusions are mainly detected within oligodendrocytes named glial cytoplasmic inclusions (GCIs). The presence of pathologically aggregated α-synuclein along with components of the protein degradation machinery, such as ubiquitin and p62, in LBs and GCIs is considered to underlie the pathogenic cascade that eventually leads to the severe neurodegeneration and neuroinflammation that characterizes these diseases. Importantly, α-synuclein is proposed to undergo pathogenic misfolding and oligomerization into higher-order structures, revealing self-templating conformations, and to exert the ability of "" spreading between cells. Therefore, the manner in which the protein is produced, is modified within neural cells and is degraded, represents a major focus of current research efforts in the field. Given that α-synuclein protein load is critical to disease pathogenesis, the identification of means to limit intracellular protein burden and halt α-synuclein propagation represents an obvious therapeutic approach in synucleinopathies. However, up to date the development of effective therapeutic strategies to prevent degeneration in synucleinopathies is limited, due to the lack of knowledge regarding the precise mechanisms underlying the observed pathology. This review critically summarizes the recent developed strategies to counteract α-synuclein toxicity, including those aimed to increase protein degradation, to prevent protein aggregation and cell-to-cell propagation, or to engage antibodies against α-synuclein and discuss open questions and unknowns for future therapeutic approaches.
帕金森病(PD)、多系统萎缩(MSA)和路易体痴呆(DLB)是病理上相似的进行性神经退行性疾病,其特征是神经元蛋白α-突触核蛋白发生病理性聚集。PD和DLB的特征是α-突触核蛋白在神经元内的蛋白质包涵体(称为路易小体[LBs]和路易神经突[LNs])中异常积累和聚集,而在MSA中,α-突触核蛋白包涵体主要在少突胶质细胞内检测到,称为胶质细胞质包涵体(GCIs)。在LBs和GCIs中,病理性聚集的α-突触核蛋白与蛋白质降解机制的成分(如泛素和p62)同时存在,被认为是导致最终严重神经退行性变和神经炎症的致病级联反应的基础,而这些正是这些疾病的特征。重要的是,有人提出α-突触核蛋白会发生致病性错误折叠并寡聚成高阶结构,呈现自我模板化构象,并具有在细胞间“传播”的能力。因此,该蛋白的产生方式、在神经细胞内的修饰方式以及降解方式,是该领域当前研究工作的主要重点。鉴于α-突触核蛋白的蛋白负荷对疾病发病机制至关重要,确定限制细胞内蛋白负荷并阻止α-突触核蛋白传播的方法,显然是突触核蛋白病的一种治疗方法。然而,由于缺乏对所观察到的病理背后精确机制的了解,迄今为止,预防突触核蛋白病退变的有效治疗策略的开发仍然有限。本综述批判性地总结了最近开发的对抗α-突触核蛋白毒性的策略,包括那些旨在增加蛋白降解、防止蛋白聚集和细胞间传播,或使用抗α-突触核蛋白抗体的策略,并讨论了未来治疗方法的开放性问题和未知因素。