Sohpal Vipan Kumar
Department of Chemical & Bio Engineering, Beant College of Engineering & Technology, Gurdaspur 143521, India.
Genomics Inform. 2020 Sep;18(3):e30. doi: 10.5808/GI.2020.18.3.e30. Epub 2020 Sep 24.
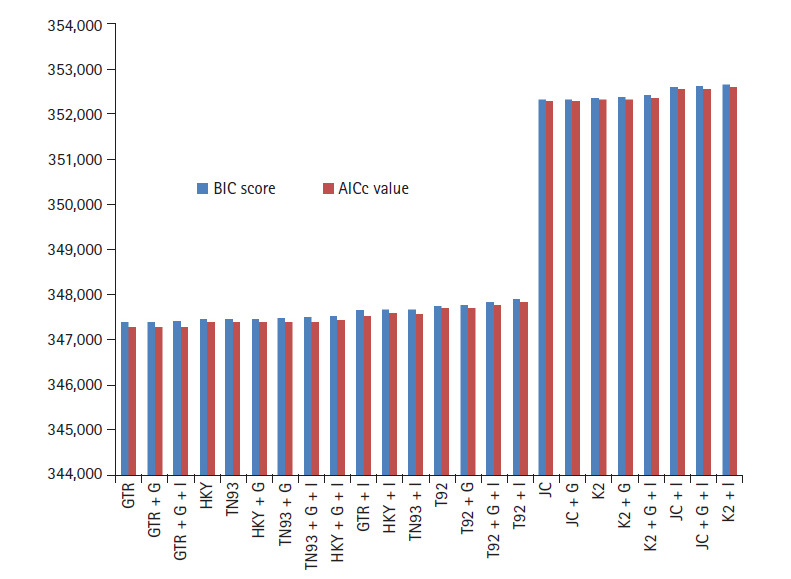
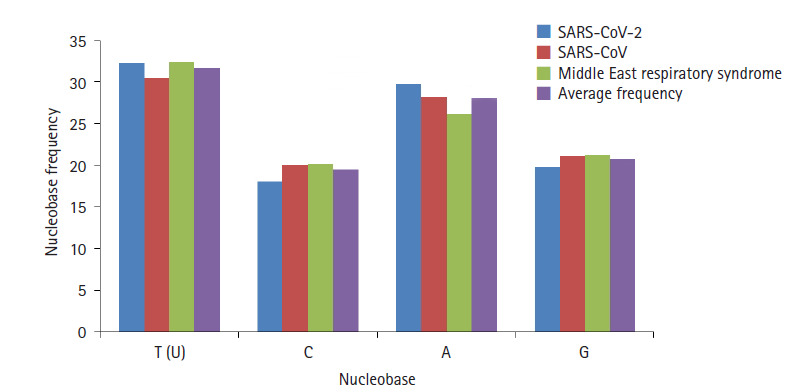
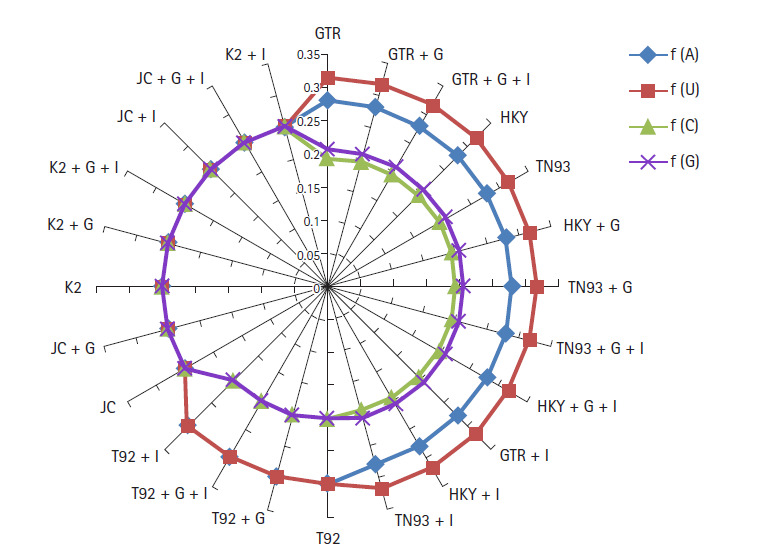
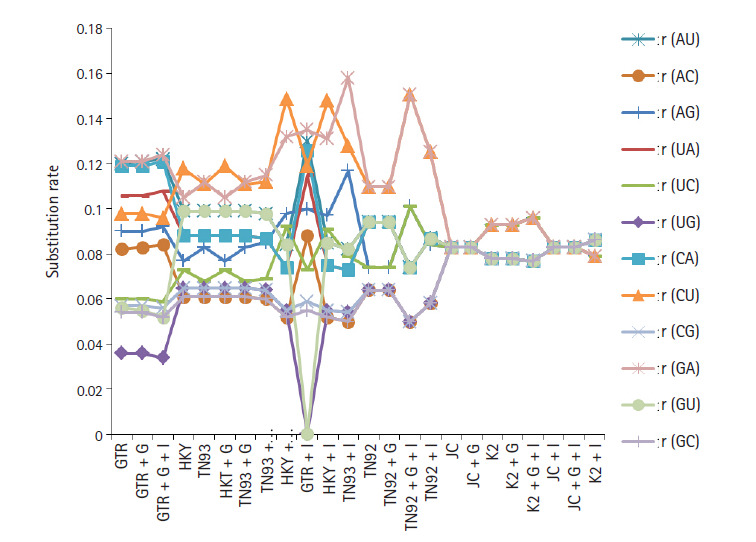
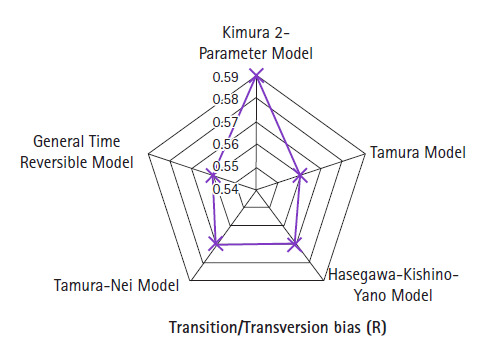
The novel coronavirus pandemic that has originated from China and spread throughout the world in three months. Genome of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) predecessor, severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) play an important role in understanding the concept of genetic variation. In this paper, the genomic data accessed from National Center for Biotechnology Information (NCBI) through Molecular Evolutionary Genetic Analysis (MEGA) for statistical analysis. Firstly, the Bayesian information criterion (BIC) and Akaike information criterion (AICc) are used to evaluate the best substitution pattern. Secondly, the maximum likelihood method used to estimate of transition/transversions (R) through Kimura-2, Tamura-3, Hasegawa-Kishino-Yano, and Tamura-Nei nucleotide substitutions model. Thirdly and finally nucleotide frequencies computed based on genomic data of NCBI. The results indicate that general times reversible model has the lowest BIC and AICc score 347,394 and 347,287, respectively. The transition/transversions bias for nucleotide substitutions models varies from 0.56 to 0.59 in MEGA output. The average nitrogenous bases frequency of U, C, A, and G are 31.74, 19.48, 28.04, and 20.74, respectively in percentages. Overall the genomic data analysis of SARS-CoV-2, SARS-CoV, and MERS-CoV highlights the close genetic relationship.
源自中国并在三个月内蔓延至全球的新型冠状病毒大流行。严重急性呼吸综合征冠状病毒2(SARS-CoV-2)的前身严重急性呼吸综合征冠状病毒(SARS-CoV)和中东呼吸综合征冠状病毒(MERS-CoV)的基因组在理解基因变异概念方面发挥着重要作用。在本文中,通过分子进化遗传分析(MEGA)从美国国立生物技术信息中心(NCBI)获取基因组数据进行统计分析。首先,使用贝叶斯信息准则(BIC)和赤池信息准则(AICc)来评估最佳替代模式。其次,使用最大似然法通过Kimura-2、Tamura-3、Hasegawa-Kishino-Yano和Tamura-Nei核苷酸替代模型来估计转换/颠换(R)。第三也是最后,根据NCBI的基因组数据计算核苷酸频率。结果表明,通用时间可逆模型的BIC和AICc得分最低,分别为347,394和347,287。在MEGA输出中,核苷酸替代模型的转换/颠换偏差在0.56至0.59之间变化。U、C、A和G的平均含氮碱基频率分别为31.74%、19.48%、28.04%和20.74%。总体而言,对SARS-CoV-2、SARS-CoV和MERS-CoV的基因组数据分析突出了它们密切的遗传关系。