Department of Molecular, Cell and Developmental Biology, University of California at Los Angeles, Los Angeles, CA, 90095, USA.
Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research, University of California at Los Angeles, Los Angeles, CA, 90095, USA.
Epigenetics Chromatin. 2020 Oct 7;13(1):42. doi: 10.1186/s13072-020-00361-9.
5' methylation of cytosines in DNA molecules is an important epigenetic mark in eukaryotes. Bisulfite sequencing is the gold standard of DNA methylation detection, and whole-genome bisulfite sequencing (WGBS) has been widely used to detect methylation at single-nucleotide resolution on a genome-wide scale. However, sodium bisulfite is known to severely degrade DNA, which, in combination with biases introduced during PCR amplification, leads to unbalanced base representation in the final sequencing libraries. Enzymatic conversion of unmethylated cytosines to uracils can achieve the same end product for sequencing as does bisulfite treatment and does not affect the integrity of the DNA; enzymatic methylation sequencing may, thus, provide advantages over bisulfite sequencing.
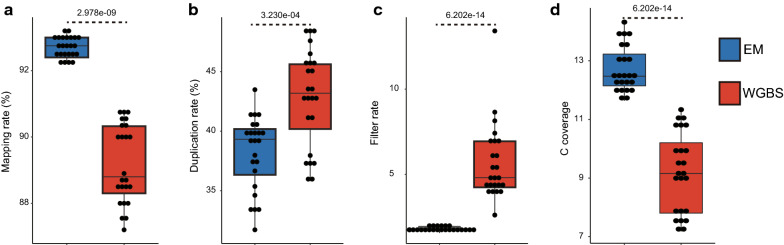
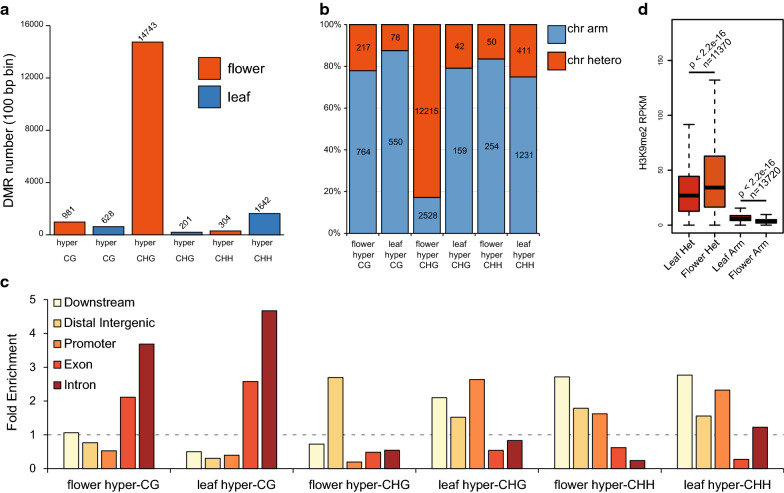
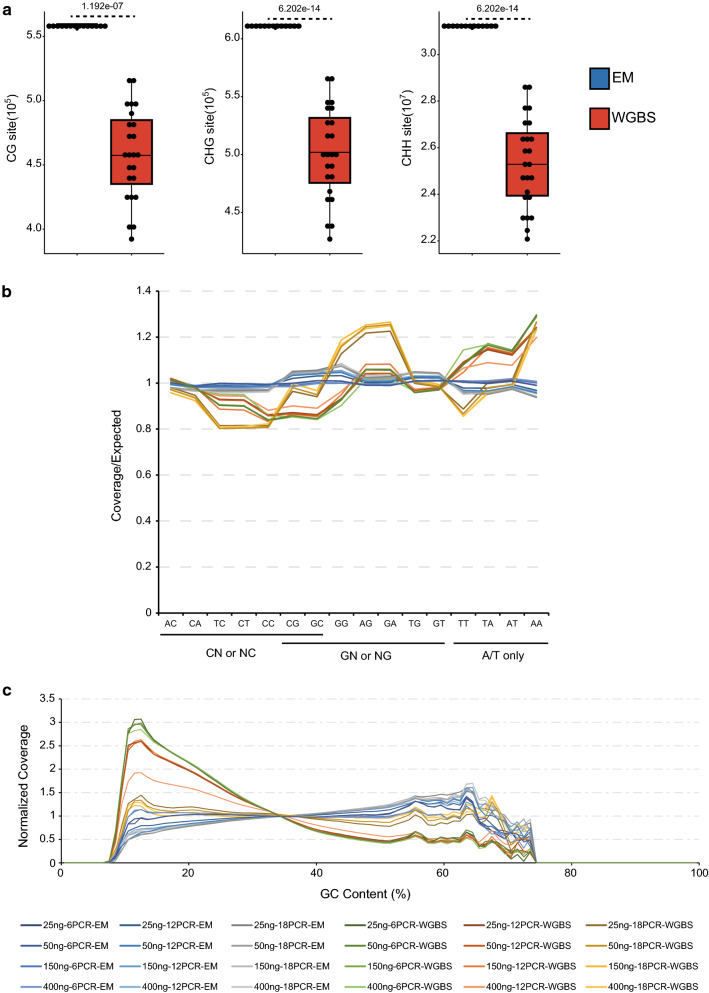
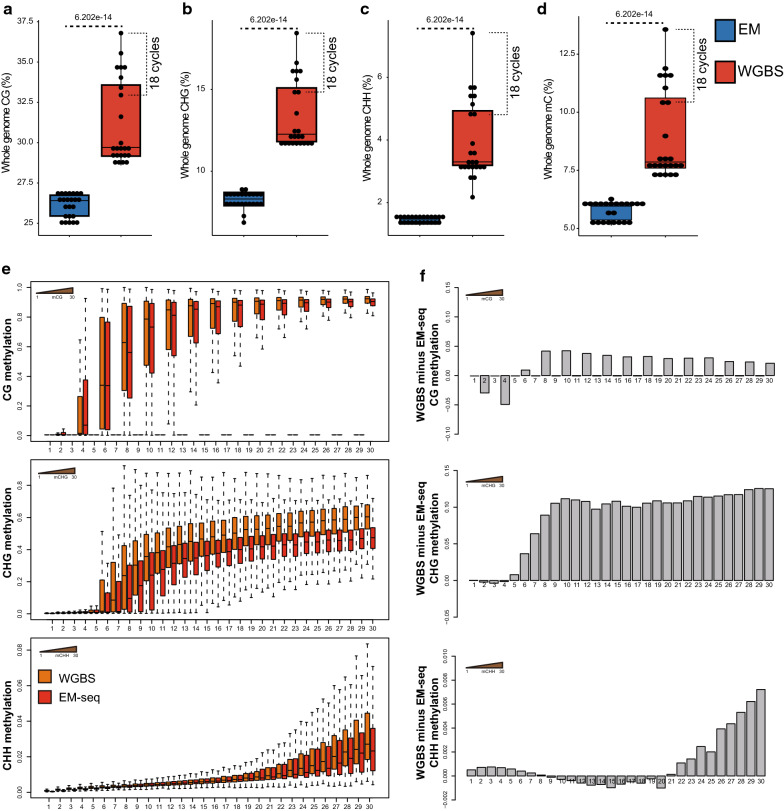
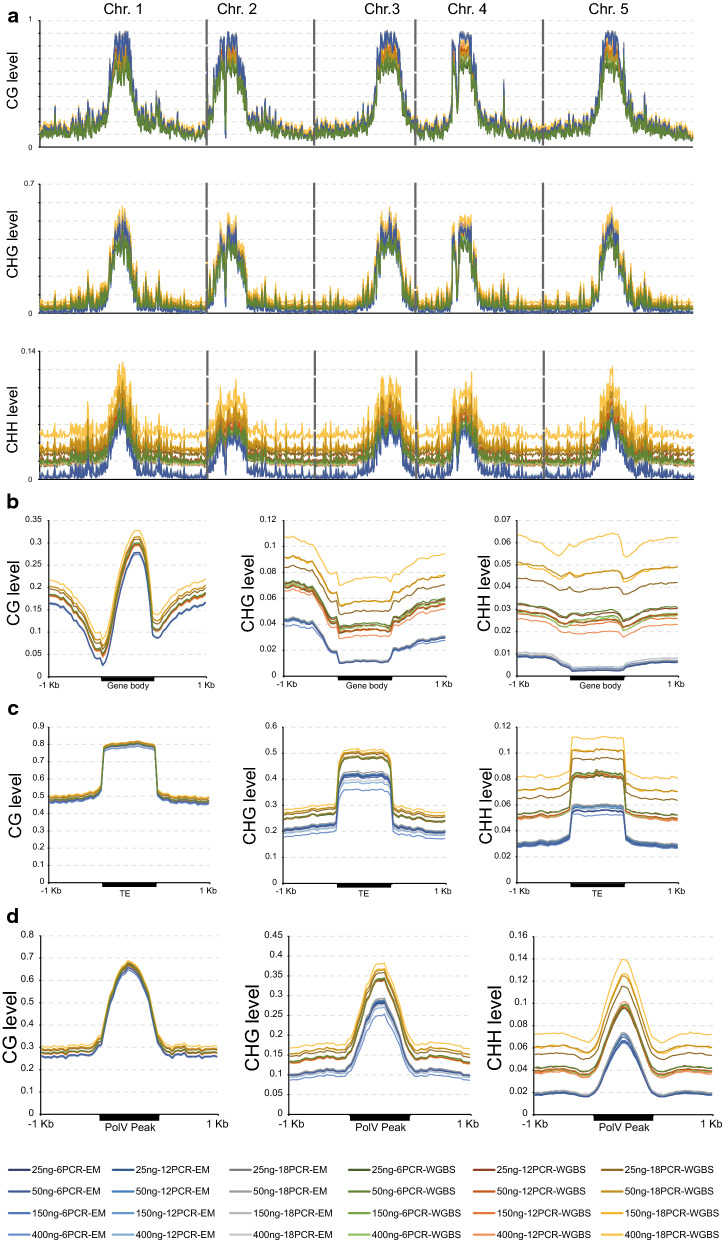
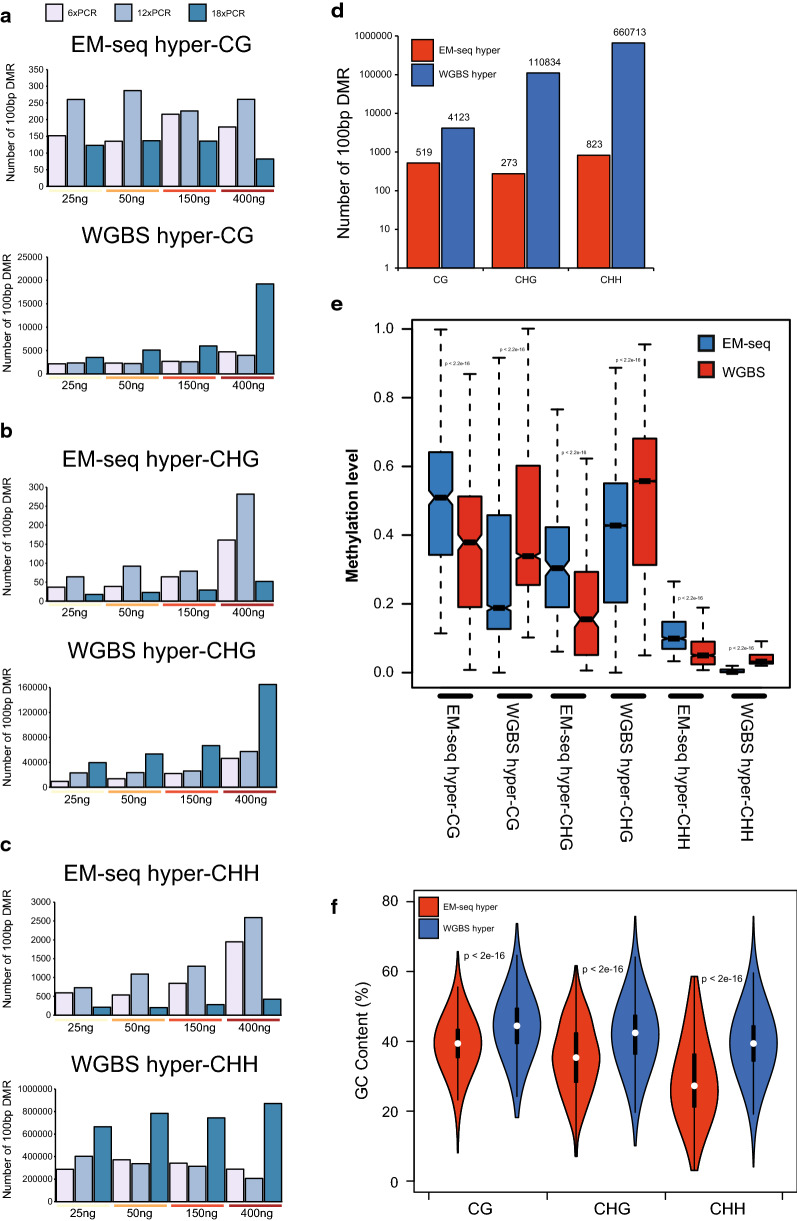
Using an enzymatic methyl-seq (EM-seq) technique to selectively deaminate unmethylated cytosines to uracils, we generated and sequenced libraries based on different amounts of Arabidopsis input DNA and different numbers of PCR cycles, and compared these data to results from traditional whole-genome bisulfite sequencing. We found that EM-seq libraries were more consistent between replicates and had higher mapping and lower duplication rates, lower background noise, higher average coverage, and higher coverage of total cytosines. Differential methylation region (DMR) analysis showed that WGBS tended to over-estimate methylation levels especially in CHG and CHH contexts, whereas EM-seq detected higher CG methylation levels in certain highly methylated areas. These phenomena can be mostly explained by a correlation of WGBS methylation estimation with GC content and methylated cytosine density. We used EM-seq to compare methylation between leaves and flowers, and found that CHG methylation level is greatly elevated in flowers, especially in pericentromeric regions.
We suggest that EM-seq is a more accurate and reliable approach than WGBS to detect methylation. Compared to WGBS, the results of EM-seq are less affected by differences in library preparation conditions or by the skewed base composition in the converted DNA. It may therefore be more desirable to use EM-seq in methylation studies.
DNA 分子中胞嘧啶的 5' 甲基化是真核生物中重要的表观遗传标记。亚硫酸氢盐测序是 DNA 甲基化检测的金标准,全基因组亚硫酸氢盐测序(WGBS)已广泛用于在全基因组范围内以单核苷酸分辨率检测甲基化。然而,亚硫酸氢钠已知会严重降解 DNA,再加上 PCR 扩增过程中引入的偏差,会导致最终测序文库中碱基的不平衡表达。未甲基化胞嘧啶的酶促转化为尿嘧啶可以达到与亚硫酸氢盐处理相同的测序终产物,并且不会影响 DNA 的完整性;因此,酶促甲基化测序可能优于亚硫酸氢盐测序。
使用酶促甲基化测序(EM-seq)技术选择性地将未甲基化的胞嘧啶脱氨为尿嘧啶,我们基于不同数量的拟南芥输入 DNA 和不同数量的 PCR 循环生成和测序文库,并将这些数据与传统的全基因组亚硫酸氢盐测序结果进行比较。我们发现,EM-seq 文库在重复之间更一致,具有更高的映射率和更低的重复率、更低的背景噪声、更高的平均覆盖率和更高的总胞嘧啶覆盖率。差异甲基化区域(DMR)分析表明,WGBS 往往会高估甲基化水平,尤其是在 CHG 和 CHH 背景下,而 EM-seq 在某些高度甲基化区域检测到更高的 CG 甲基化水平。这些现象主要可以通过 WGBS 甲基化估计与 GC 含量和甲基化胞嘧啶密度的相关性来解释。我们使用 EM-seq 比较了叶和花之间的甲基化,发现 CHG 甲基化水平在花中大大升高,尤其是在着丝粒区域。
我们建议 EM-seq 是一种比 WGBS 更准确、更可靠的检测甲基化的方法。与 WGBS 相比,EM-seq 的结果受文库制备条件差异或转化 DNA 中偏倚碱基组成的影响较小。因此,在甲基化研究中可能更希望使用 EM-seq。