Epigenetics Programme, The Babraham Institute, Cambridge, CB22 3AT, UK.
Present address: MRC Human Genetics Unit, MRC Institute of Genetics and Molecular Medicine, University of Edinburgh, Edinburgh, EH4 2XU, UK.
Genome Biol. 2018 Mar 15;19(1):33. doi: 10.1186/s13059-018-1408-2.
Whole-genome bisulfite sequencing (WGBS) is becoming an increasingly accessible technique, used widely for both fundamental and disease-oriented research. Library preparation methods benefit from a variety of available kits, polymerases and bisulfite conversion protocols. Although some steps in the procedure, such as PCR amplification, are known to introduce biases, a systematic evaluation of biases in WGBS strategies is missing.
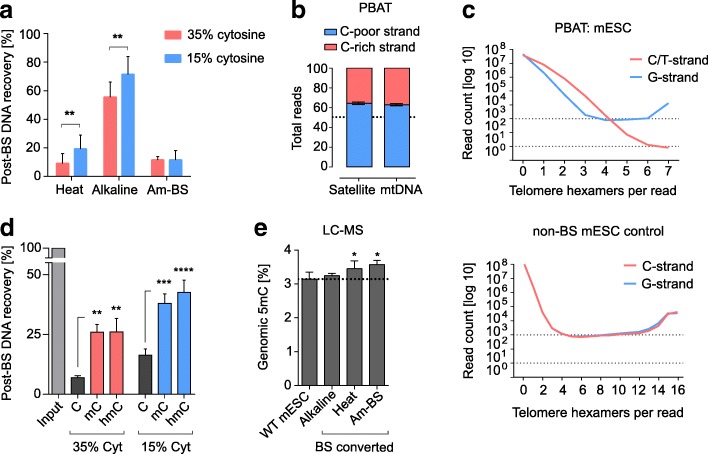
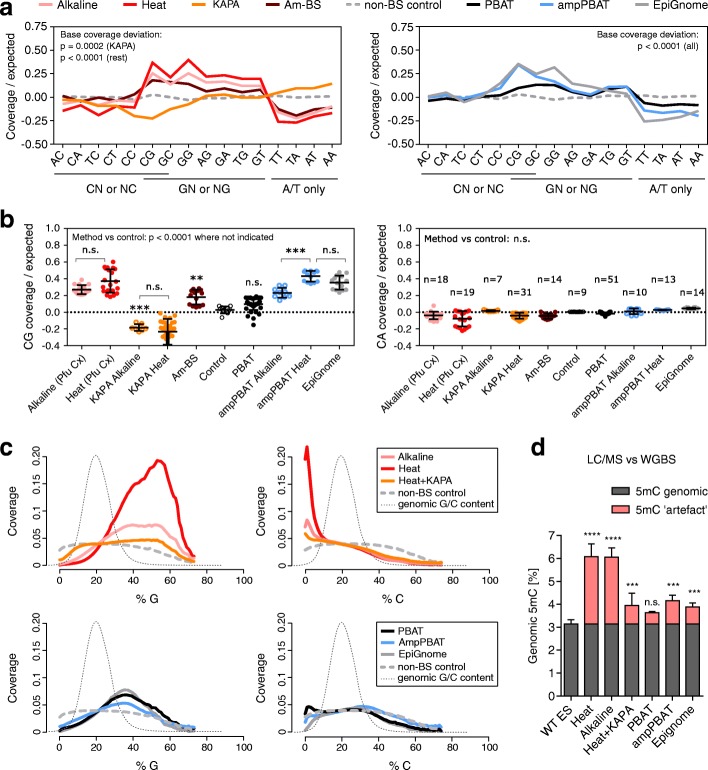
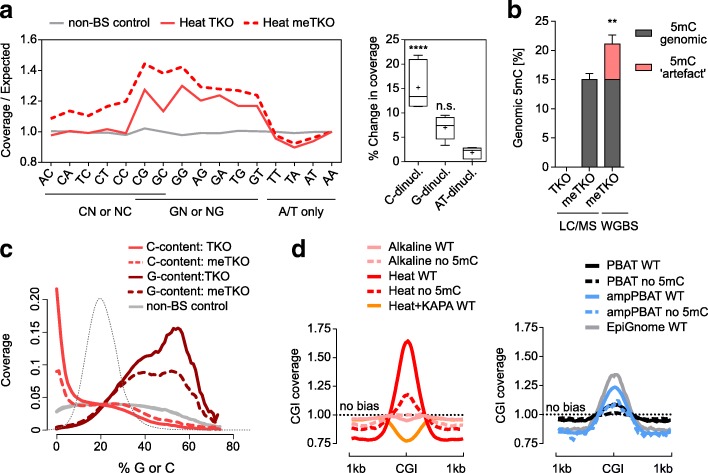
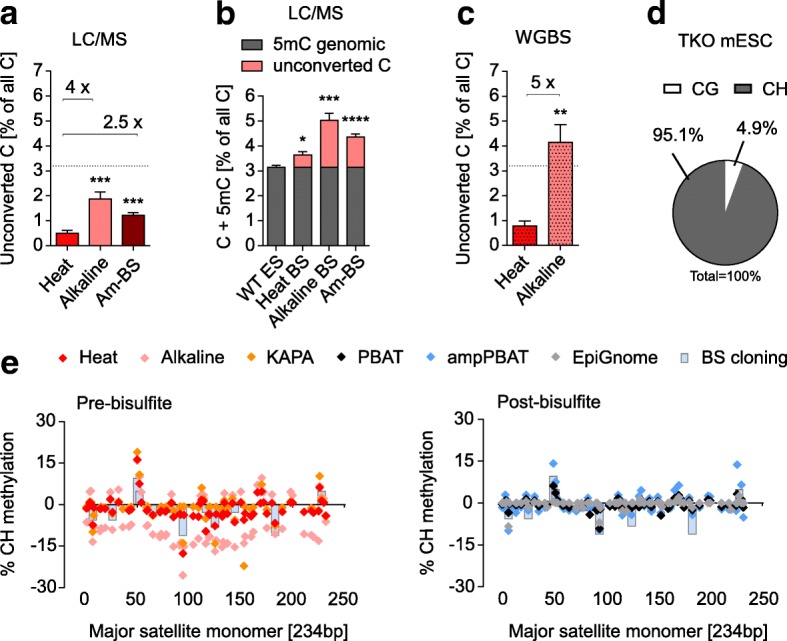
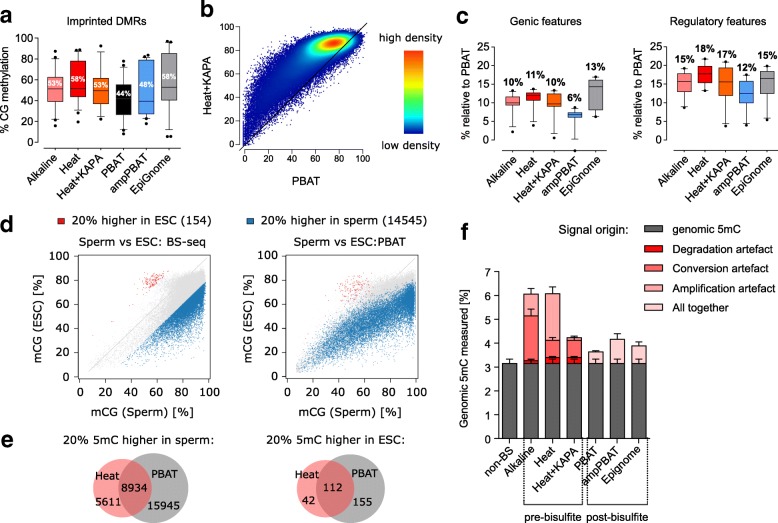
We perform a comparative analysis of several commonly used pre- and post-bisulfite WGBS library preparation protocols for their performance and quality of sequencing outputs. Our results show that bisulfite conversion per se is the main trigger of pronounced sequencing biases, and PCR amplification builds on these underlying artefacts. The majority of standard library preparation methods yield a significantly biased sequence output and overestimate global methylation. Importantly, both absolute and relative methylation levels at specific genomic regions vary substantially between methods, with clear implications for DNA methylation studies.
We show that amplification-free library preparation is the least biased approach for WGBS. In protocols with amplification, the choice of bisulfite conversion protocol or polymerase can significantly minimize artefacts. To aid with the quality assessment of existing WGBS datasets, we have integrated a bias diagnostic tool in the Bismark package and offer several approaches for consideration during the preparation and analysis of WGBS datasets.
全基因组亚硫酸氢盐测序(WGBS)技术日益普及,广泛应用于基础研究和疾病导向研究。文库制备方法受益于各种可用试剂盒、聚合酶和亚硫酸氢盐转化方案。尽管该过程中的一些步骤(如 PCR 扩增)已知会产生偏倚,但 WGBS 策略中的偏倚系统评估仍存在空白。
我们对几种常用的亚硫酸氢盐处理前后的 WGBS 文库制备方案进行了比较分析,以评估其性能和测序结果的质量。我们的结果表明,亚硫酸氢盐转化本身是引起显著测序偏倚的主要原因,而 PCR 扩增则建立在这些潜在的人为因素之上。大多数标准文库制备方法会产生明显偏倚的序列输出,并高估整体甲基化水平。重要的是,特定基因组区域的绝对和相对甲基化水平在不同方法之间存在显著差异,这对 DNA 甲基化研究具有重要影响。
我们表明,无扩增文库制备是 WGBS 中最少偏倚的方法。在具有扩增的方案中,亚硫酸氢盐转化方案或聚合酶的选择可以显著最小化人为因素。为了帮助评估现有的 WGBS 数据集的质量,我们在 Bismark 软件包中集成了一个偏倚诊断工具,并提供了几种在 WGBS 数据集制备和分析过程中考虑的方法。