National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University, Wuhan, 430070, China.
Agricultural Bioinformatics Key Laboratory of Hubei Province, Hubei Engineering Technology Research Center of Agricultural Big Data, 3D Genomics Research Center, College of Informatics, Huazhong Agricultural University, Wuhan, 430070, China.
BMC Bioinformatics. 2020 Oct 12;21(1):451. doi: 10.1186/s12859-020-03798-7.
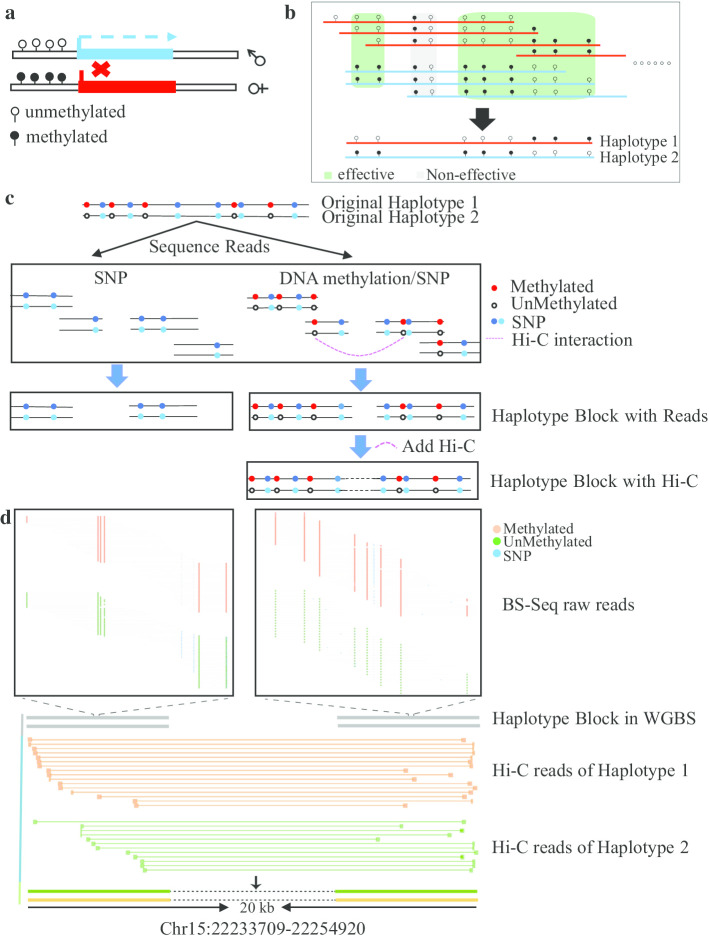
DNA methylation is an important epigenetic modification that plays a critical role in most eukaryotic organisms. Parental alleles in haploid genomes may exhibit different methylation patterns, which can lead to different phenotypes and even different therapeutic and drug responses to diseases. However, to our knowledge, no software is available for the identification of DNA methylation haplotype regions with combined allele-specific DNA methylation, single nucleotide polymorphisms (SNPs) and high-throughput chromosome conformation capture (Hi-C) data.
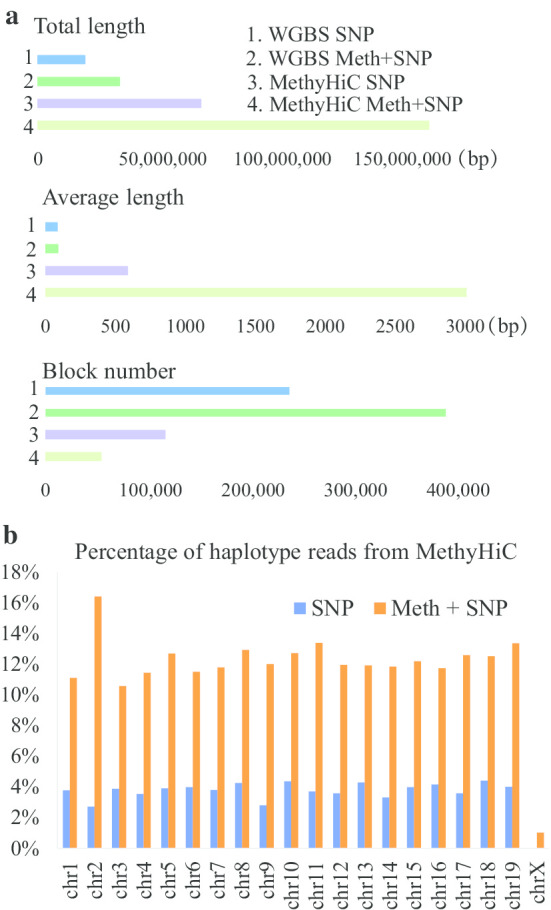
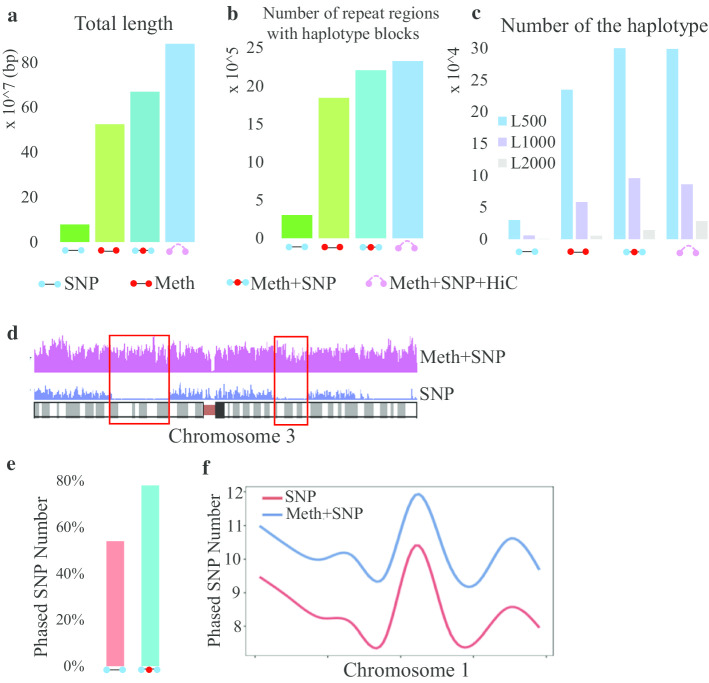
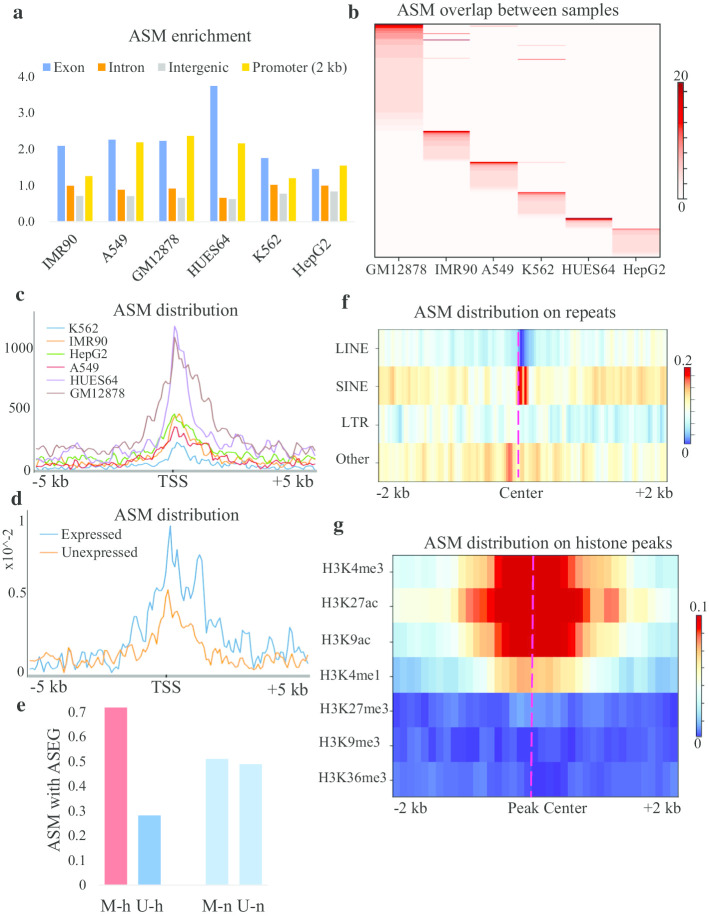
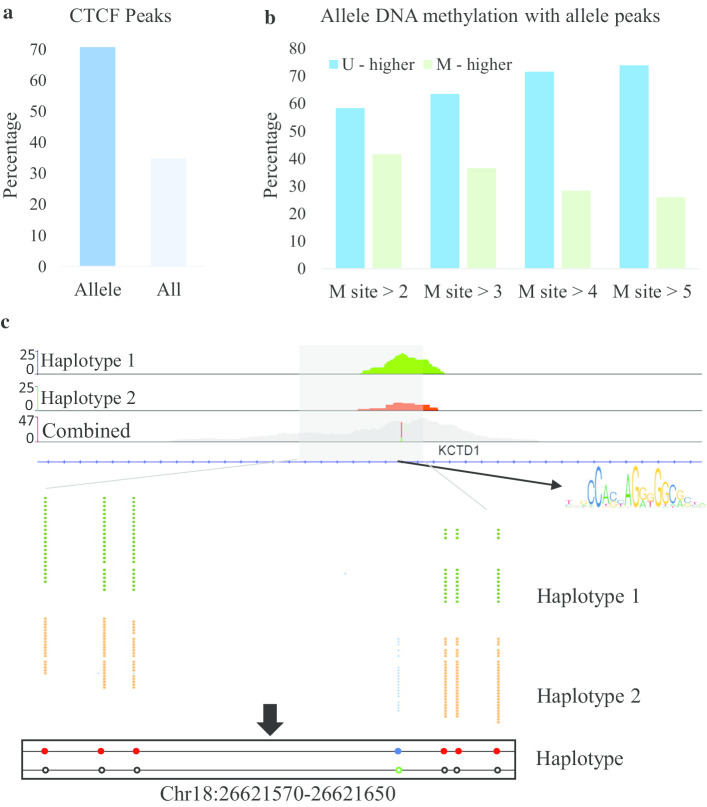
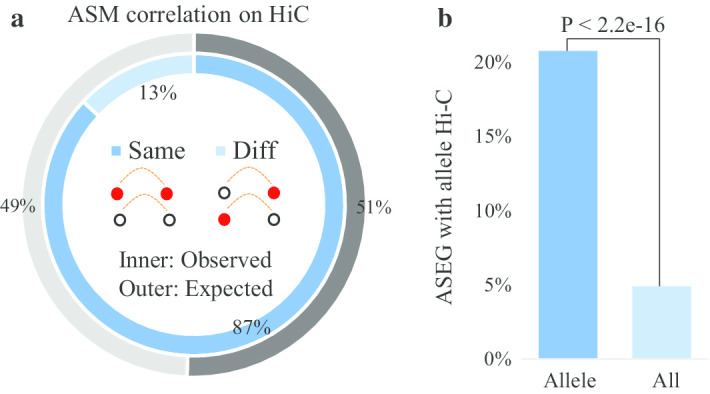
In this paper, we developed a new method, MethHaplo, that identify DNA methylation haplotype regions with allele-specific DNA methylation and SNPs from whole-genome bisulfite sequencing (WGBS) data. Our results showed that methylation haplotype regions were ten times longer than haplotypes with SNPs only. When we integrate WGBS and Hi-C data, MethHaplo could call even longer haplotypes.
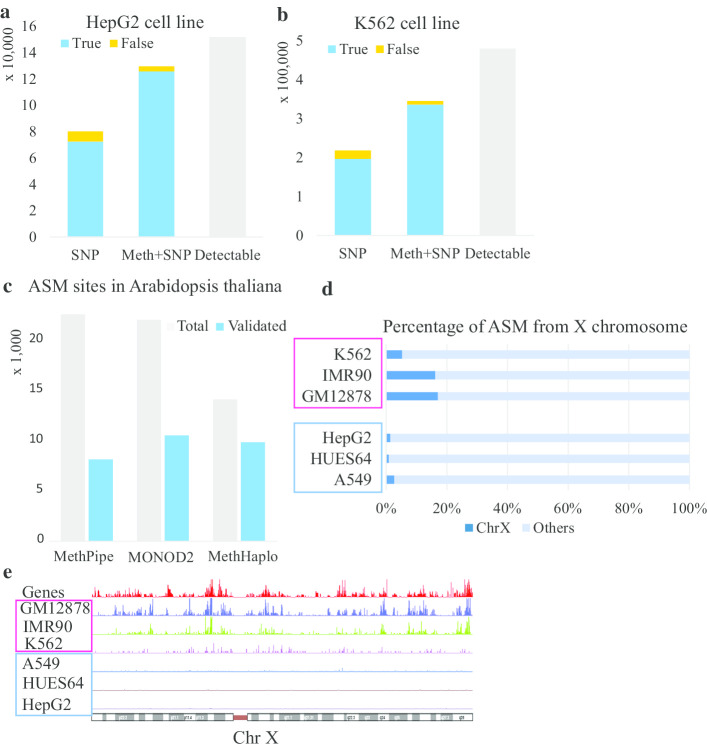
This study illustrates the usefulness of methylation haplotypes. By constructing methylation haplotypes for various cell lines, we provide a clearer picture of the effect of DNA methylation on gene expression, histone modification and three-dimensional chromosome structure at the haplotype level. Our method could benefit the study of parental inheritance-related disease and hybrid vigor in agriculture.
DNA 甲基化是一种重要的表观遗传修饰,在大多数真核生物中起着关键作用。在单倍体基因组中,亲本等位基因可能表现出不同的甲基化模式,这可能导致不同的表型,甚至对疾病的治疗和药物反应也不同。然而,据我们所知,目前还没有软件可用于识别具有组合等位基因特异性 DNA 甲基化、单核苷酸多态性(SNPs)和高通量染色体构象捕获(Hi-C)数据的 DNA 甲基化单倍型区域。
在本文中,我们开发了一种新方法 MethHaplo,可从全基因组亚硫酸氢盐测序(WGBS)数据中识别具有等位基因特异性 DNA 甲基化和 SNPs 的 DNA 甲基化单倍型区域。我们的结果表明,甲基化单倍型区域比仅具有 SNPs 的单倍型长十倍。当我们整合 WGBS 和 Hi-C 数据时,MethHaplo 甚至可以调用更长的单倍型。
这项研究说明了甲基化单倍型的有用性。通过构建各种细胞系的甲基化单倍型,我们在单倍型水平上更清楚地了解 DNA 甲基化对基因表达、组蛋白修饰和三维染色体结构的影响。我们的方法可以使与亲本遗传相关的疾病和农业杂种优势的研究受益。