Department of Psychiatry and Behavioral Sciences, Stanford University School of Medicine, Stanford, CA 94305, USA.
Department of Genetics, Stanford University School of Medicine, Stanford, CA 94305, USA.
Nucleic Acids Res. 2019 May 7;47(8):3846-3861. doi: 10.1093/nar/gkz169.
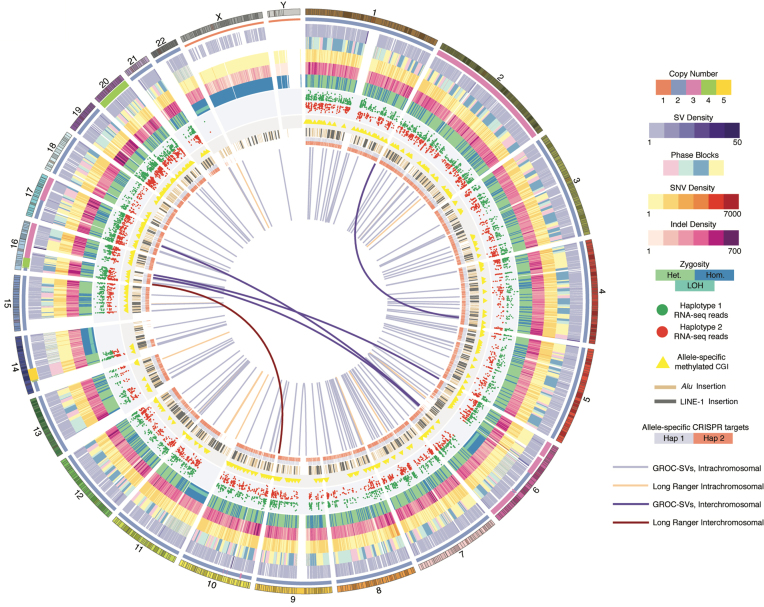
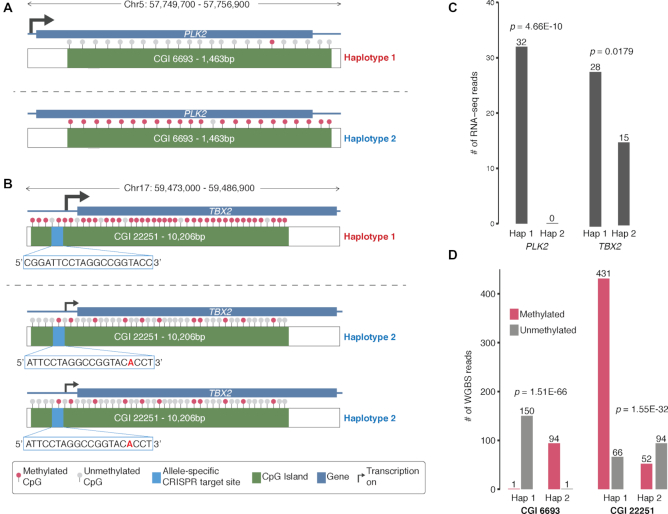
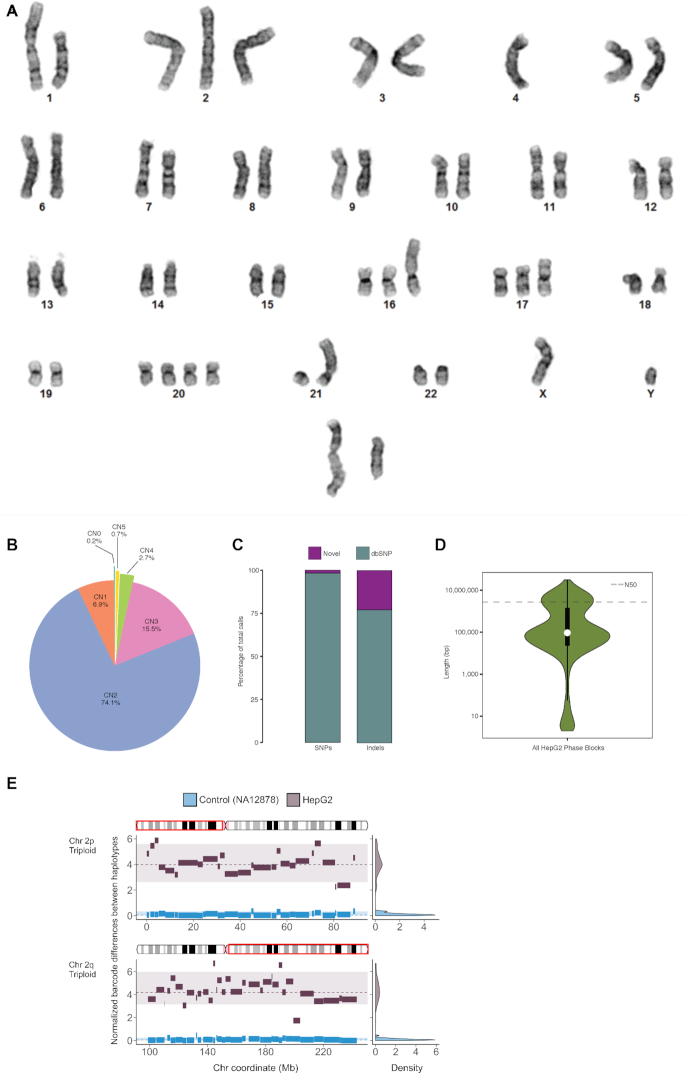
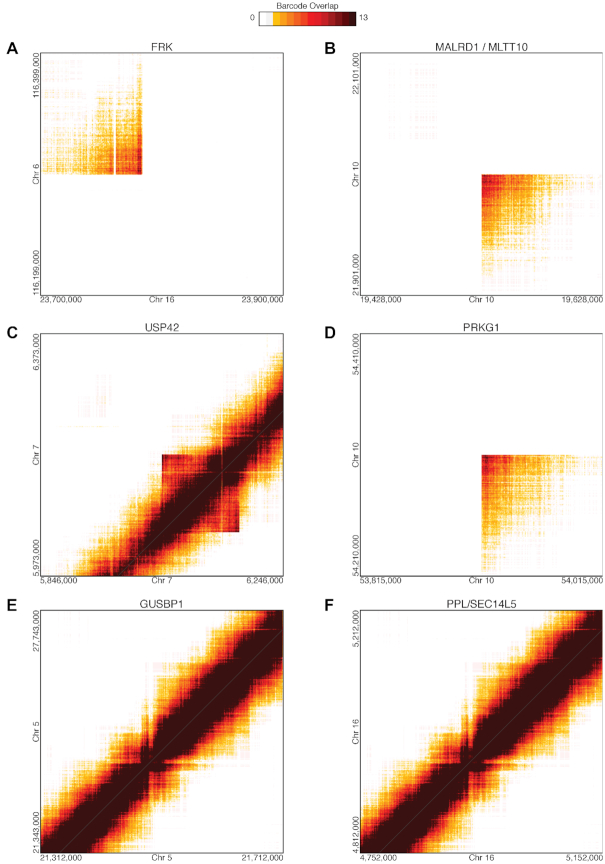
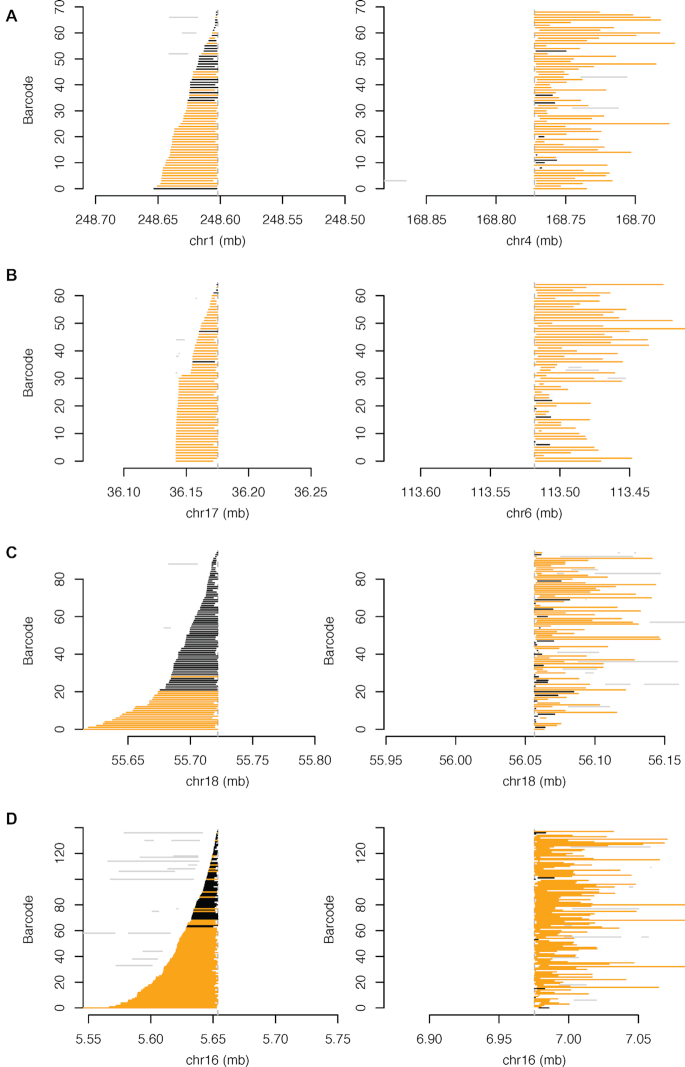
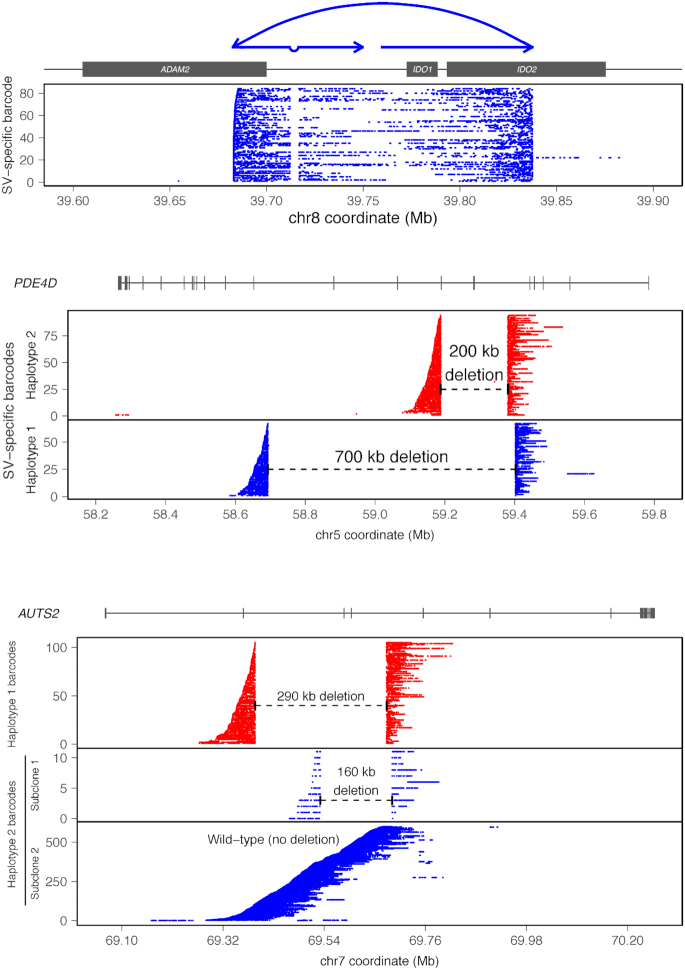
HepG2 is one of the most widely used human cancer cell lines in biomedical research and one of the main cell lines of ENCODE. Although the functional genomic and epigenomic characteristics of HepG2 are extensively studied, its genome sequence has never been comprehensively analyzed and higher order genomic structural features are largely unknown. The high degree of aneuploidy in HepG2 renders traditional genome variant analysis methods challenging and partially ineffective. Correct and complete interpretation of the extensive functional genomics data from HepG2 requires an understanding of the cell line's genome sequence and genome structure. Using a variety of sequencing and analysis methods, we identified a wide spectrum of genome characteristics in HepG2: copy numbers of chromosomal segments at high resolution, SNVs and Indels (corrected for aneuploidy), regions with loss of heterozygosity, phased haplotypes extending to entire chromosome arms, retrotransposon insertions and structural variants (SVs) including complex and somatic genomic rearrangements. A large number of SVs were phased, sequence assembled and experimentally validated. We re-analyzed published HepG2 datasets for allele-specific expression and DNA methylation and assembled an allele-specific CRISPR/Cas9 targeting map. We demonstrate how deeper insights into genomic regulatory complexity are gained by adopting a genome-integrated framework.
HepG2 是人癌细胞系中应用最广泛的细胞系之一,也是 ENCODE 的主要细胞系之一。尽管 HepG2 的功能基因组学和表观基因组学特征得到了广泛的研究,但它的基因组序列从未被全面分析过,其高级别基因组结构特征也在很大程度上是未知的。HepG2 的高度非整倍性使得传统的基因组变异分析方法具有挑战性且部分无效。要正确完整地解释来自 HepG2 的广泛的功能基因组学数据,就需要了解细胞系的基因组序列和基因组结构。我们使用多种测序和分析方法,鉴定出 HepG2 具有广泛的基因组特征:染色体片段的高分辨率拷贝数、SNVs 和 Indels(针对非整倍性进行了校正)、杂合性丢失区域、扩展到整个染色体臂的相定单倍型、逆转录转座子插入和结构变异(SVs),包括复杂的和体细胞基因组重排。我们对大量的 SVs 进行了相定、序列组装和实验验证。我们重新分析了已发表的 HepG2 数据集,以研究等位基因特异性表达和 DNA 甲基化,并组装了等位基因特异性的 CRISPR/Cas9 靶向图谱。我们展示了通过采用基因组综合框架如何获得对基因组调控复杂性的更深入了解。