Institut de recherches cliniques de Montréal, IRCM, Montréal, QC, Canada.
Dept. of Pathology, Biochemistry and Molecular Biology, Dalhousie University, Halifax, NS, Canada.
Haematologica. 2020 Oct 1;105(10):2457-2470. doi: 10.3324/haematol.2019.222596.
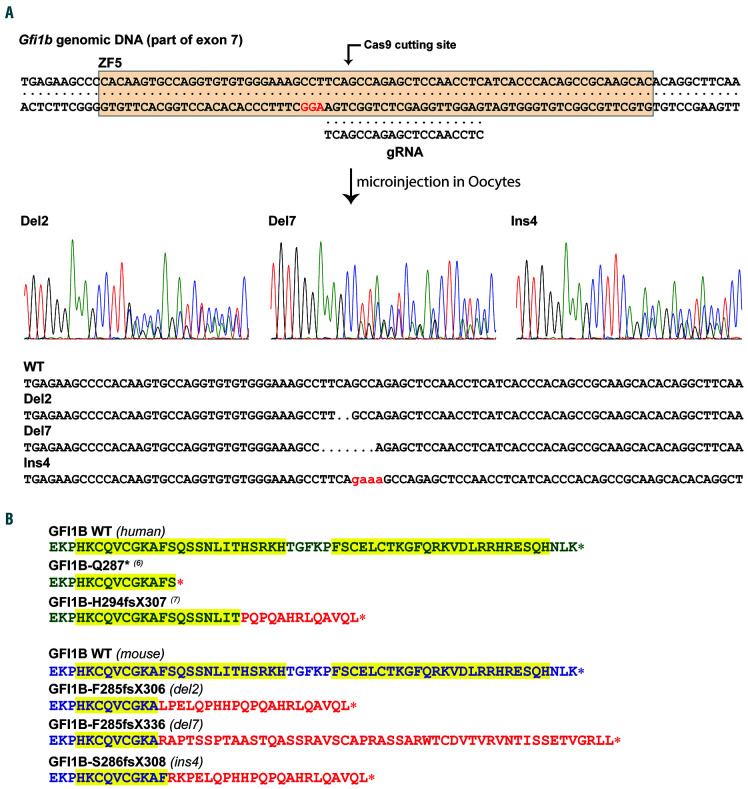
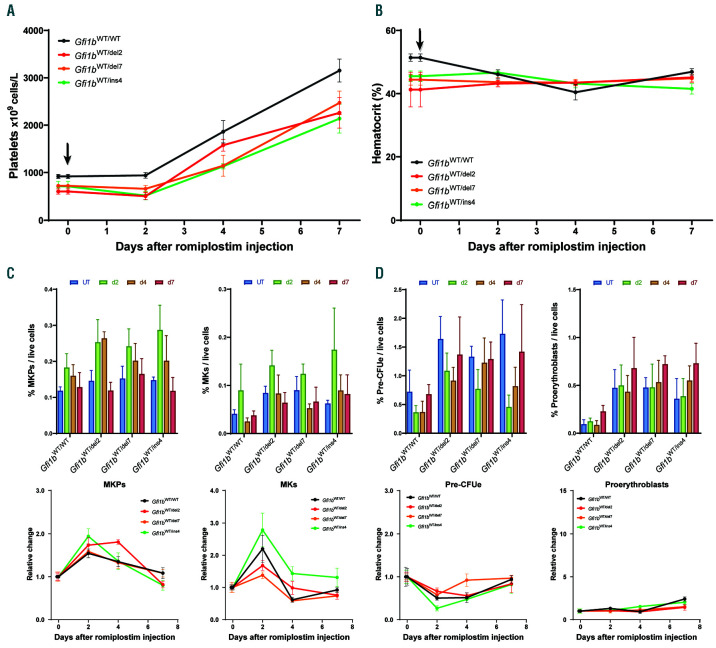
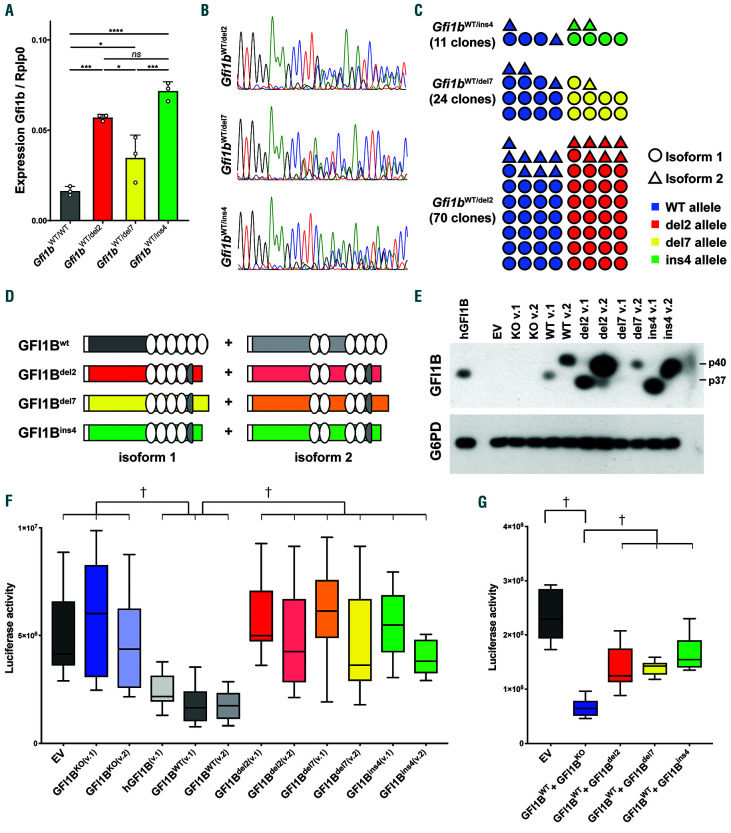
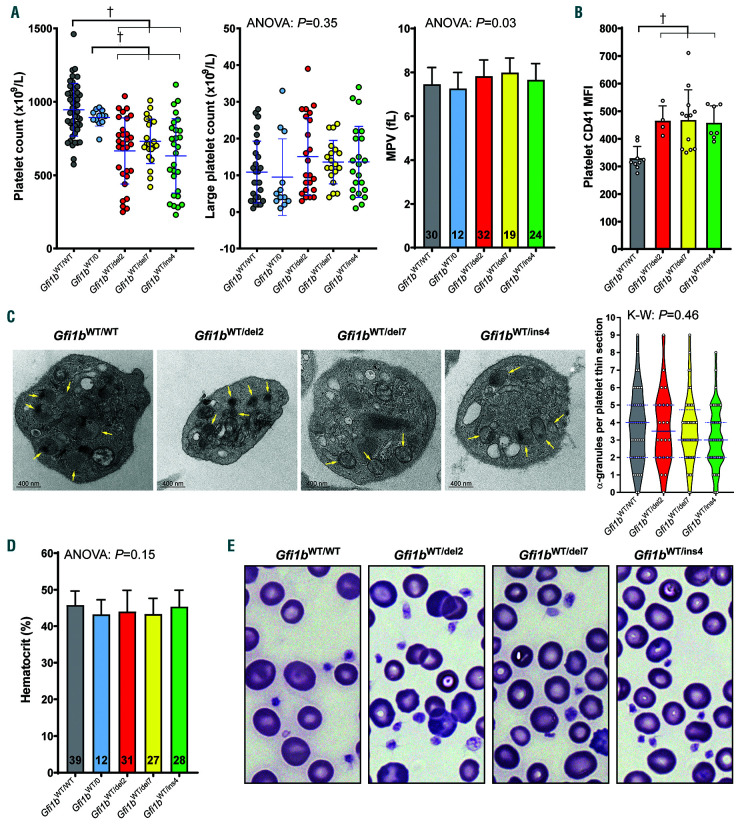
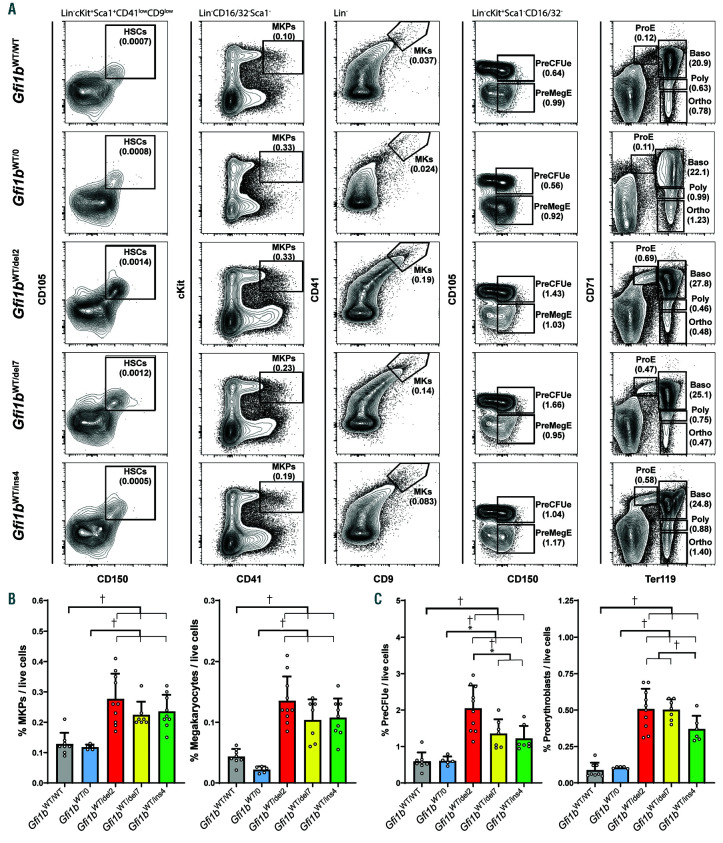
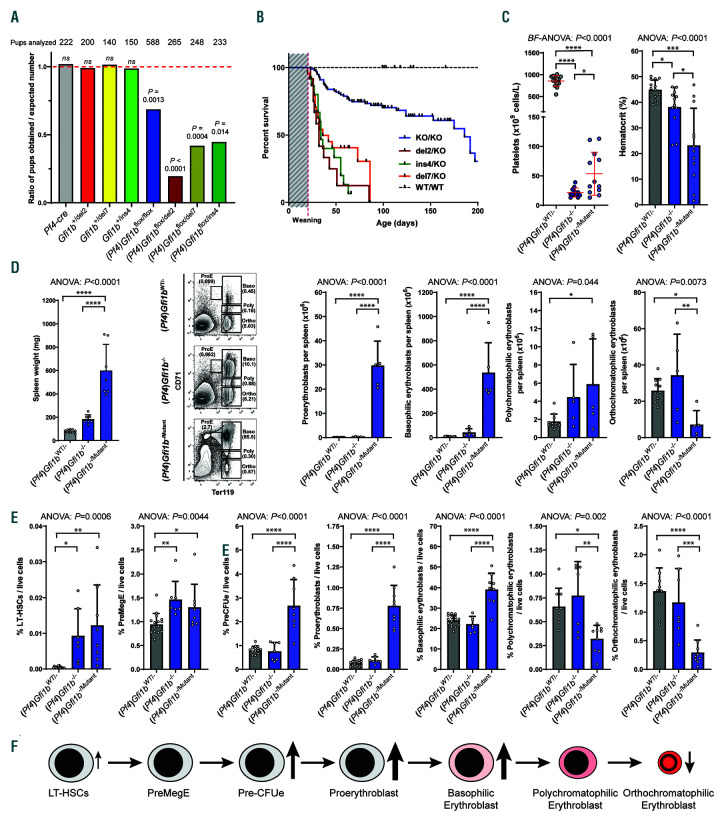
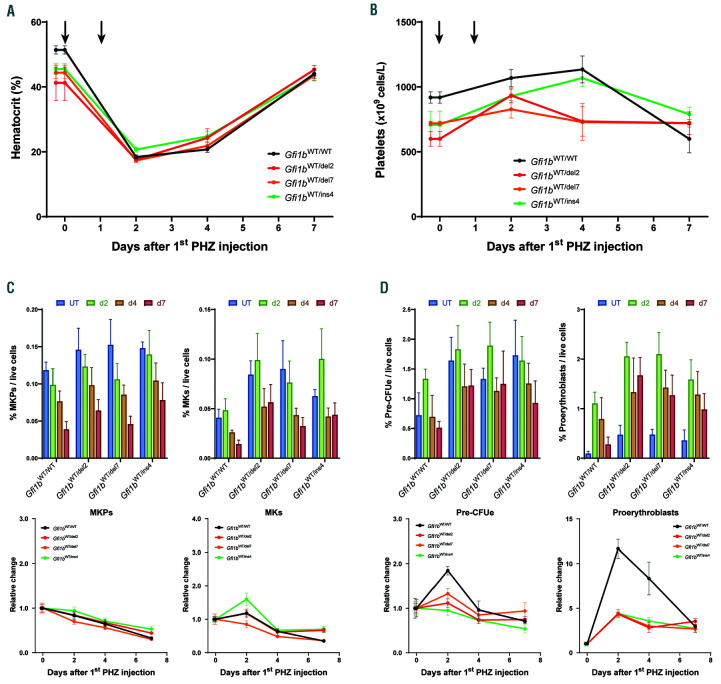
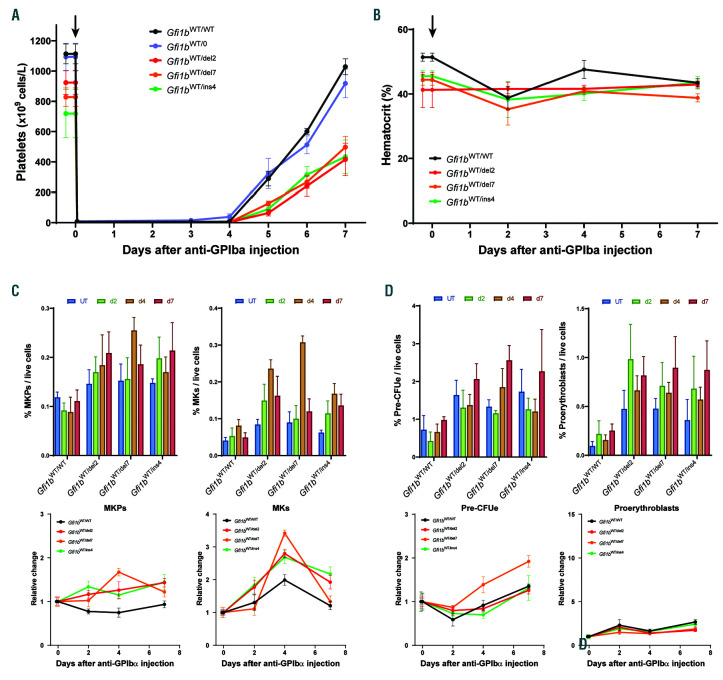
GFI1B-related thrombocytopenia (GFI1B-RT) is a rare bleeding disorder mainly caused by the presence of truncated GFI1B proteins with dominant-negative properties. The disease is characterized by low platelet counts, the presence of abnormal platelets, a megakaryocytic expansion and mild erythroid defects. However, no animal models faithfully reproducing the GFI1B-RT phenotype observed in patients exist. We had previously generated mice with floxed Gfi1b alleles that can be eliminated by Cre recombinase, but those animals developed a much more severe phenotype than GFI1B-RT patients and were of limited interest in assessing the disease. Using CRISPR/Cas9 technology, we have now established three independent mouse lines that carry mutated Gfi1b alleles producing proteins lacking DNA binding zinc fingers and thereby acting in a dominant negative (DN) manner. Mice heterozygous for these Gfi1b-DN alleles show reduced platelet counts and an expansion of megakaryocytes similar to features of human GFI1B-RT but lacking the distinctively large agranular platelets. In addition, Gfi1b-DN mice exhibit an expansion of erythroid precursors indicative of a mildly abnormal erythropoiesis but without noticeable red blood cell defects. When associated with megakaryocyte-specific ablation of the remaining allele, the Gfi1b-DN alleles triggered erythroid-specific deleterious defects. Gfi1b-DN mice also showed a delayed recovery from platelet depletion, indicating a defect in stress thrombopoiesis. However, injecting Gfi1b-DN mice with romiplostim, a thrombopoietin receptor super agonist, increased platelet numbers even beyond normal levels. Thus, our data support a causal link between DN mutations in GFI1B and thrombocytopenia and suggest that patients with GFI1B-RT could be treated successfully with thrombopoietin agonists.
GFI1B 相关血小板减少症(GFI1B-RT)是一种罕见的出血性疾病,主要由具有显性负性的截短 GFI1B 蛋白引起。该疾病的特征是血小板计数低、存在异常血小板、巨核细胞扩张和轻度红细胞缺陷。然而,目前还没有能够真实再现患者中观察到的 GFI1B-RT 表型的动物模型。我们之前曾生成了带有 floxed Gfi1b 等位基因的小鼠,这些基因可以通过 Cre 重组酶消除,但这些动物的表型比 GFI1B-RT 患者更为严重,并且在评估疾病方面的意义有限。我们使用 CRISPR/Cas9 技术,现在已经建立了三个独立的携带缺失 DNA 结合锌指的突变 Gfi1b 等位基因的小鼠系,从而以显性负性(DN)方式发挥作用。这些 Gfi1b-DN 等位基因杂合的小鼠表现出血小板计数减少和巨核细胞扩张,类似于人类 GFI1B-RT 的特征,但缺乏独特的大颗粒血小板。此外,Gfi1b-DN 小鼠还表现出红系前体细胞的扩张,表明存在轻度异常的红细胞生成,但没有明显的红细胞缺陷。当与巨核细胞特异性消除剩余等位基因相关联时,Gfi1b-DN 等位基因引发了红细胞特异性的有害缺陷。Gfi1b-DN 小鼠还显示出从血小板耗竭中恢复延迟,表明应激性血小板生成存在缺陷。然而,给 Gfi1b-DN 小鼠注射 romiplostim(一种血小板生成素受体超激动剂),甚至可以使血小板数量增加到正常水平以上。因此,我们的数据支持 GFI1B 中的 DN 突变与血小板减少之间存在因果关系,并表明 GFI1B-RT 患者可以成功地用血小板生成素激动剂治疗。