Department of Molecular Biology and Biochemistry, Simon Fraser University, Burnaby, BC, V5A1S6
Department of Molecular Biology and Biochemistry, Simon Fraser University, Burnaby, BC, V5A1S6.
eNeuro. 2020 Nov 5;7(6). doi: 10.1523/ENEURO.0176-20.2020. Print 2020 Nov/Dec.
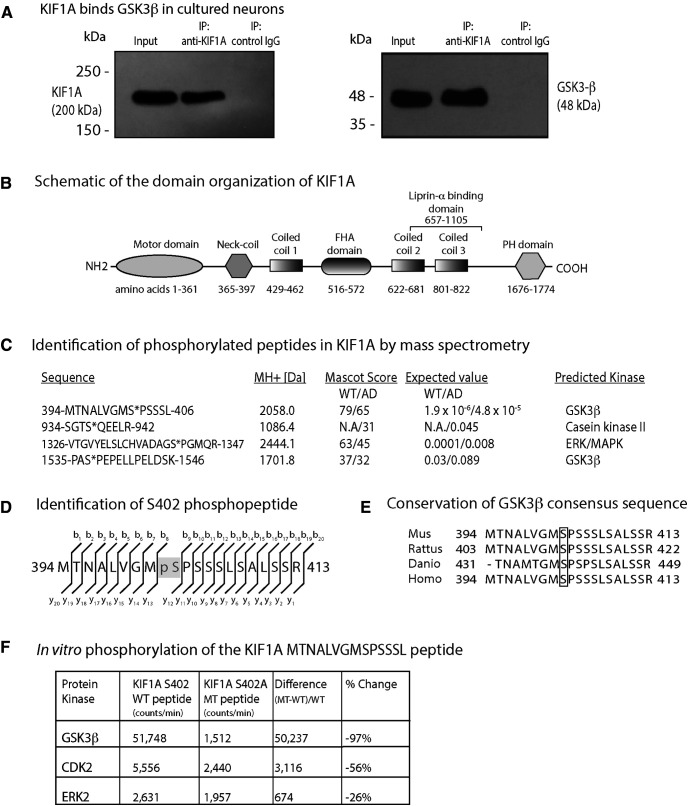
Impairment of axonal transport is an early pathologic event that precedes neurotoxicity in Alzheimer's disease (AD). Soluble amyloid-β oligomers (AβOs), a causative agent of AD, activate intracellular signaling cascades that trigger phosphorylation of many target proteins, including tau, resulting in microtubule destabilization and transport impairment. Here, we investigated how KIF1A, a kinesin-3 family motor protein required for the transport of neurotrophic factors, is impaired in mouse hippocampal neurons treated with AβOs. By live cell imaging, we observed that AβOs inhibit transport of KIF1A-GFP similarly in wild-type and tau knock-out neurons, indicating that tau is not required for this effect. Pharmacological inhibition of glycogen synthase kinase 3β (GSK3β), a kinase overactivated in AD, prevented the transport defects. By mass spectrometry on KIF1A immunoprecipitated from transgenic AD mouse brain, we detected phosphorylation at S402, which conforms to a highly conserved GSK3β consensus site. We confirmed that this site is phosphorylated by GSK3β Finally, we tested whether a phosphomimic of S402 could modulate KIF1A motility in control and AβO-treated mouse neurons and in a Golgi dispersion assay devoid of endogenous KIF1A. In both systems, transport driven by mutant motors was similar to that of WT motors. In conclusion, GSK3β impairs KIF1A transport but does not regulate motor motility at S402. Further studies are required to determine the specific phosphorylation sites on KIF1A that regulate its cargo binding and/or motility in physiological and disease states.
轴突运输的损伤是阿尔茨海默病(AD)神经毒性之前的早期病理事件。可溶性淀粉样β寡聚体(AβO)是 AD 的致病因素,它激活细胞内信号级联反应,触发许多靶蛋白(包括 tau)的磷酸化,导致微管不稳定和运输损伤。在这里,我们研究了驱动蛋白-1A(KIF1A),一种神经递质运输所必需的驱动蛋白-3 家族运动蛋白,如何在 AβO 处理的小鼠海马神经元中受到损伤。通过活细胞成像,我们观察到 AβO 抑制野生型和 tau 敲除神经元中 KIF1A-GFP 的运输,表明 tau 不是这种效应所必需的。糖原合酶激酶 3β(GSK3β)的药理学抑制,一种在 AD 中过度激活的激酶,可防止运输缺陷。通过对转 AD 小鼠脑内 KIF1A 免疫沉淀的质谱分析,我们检测到 S402 的磷酸化,这符合 GSK3β 的高度保守的共识位点。我们证实该位点由 GSK3β磷酸化。最后,我们测试了 S402 的磷酸模拟物是否可以调节对照和 AβO 处理的小鼠神经元中 KIF1A 的运动性,以及没有内源性 KIF1A 的高尔基分散测定。在这两个系统中,突变体驱动的运输与 WT 驱动的运输相似。总之,GSK3β 损伤 KIF1A 运输,但不调节 S402 的运动性。需要进一步的研究来确定 KIF1A 上调节其货物结合和/或在生理和疾病状态下运动性的特定磷酸化位点。