Department of Neurology, Mayo Clinic, 200 First St. SW, Rochester, MN 55905, USA.
Microscopy and Cell Analysis Core Facility, Mayo Clinic, 200 First St. SW, Rochester, MN, 55905, USA.
Neurobiol Dis. 2018 Jun;114:1-16. doi: 10.1016/j.nbd.2018.02.003. Epub 2018 Mar 2.
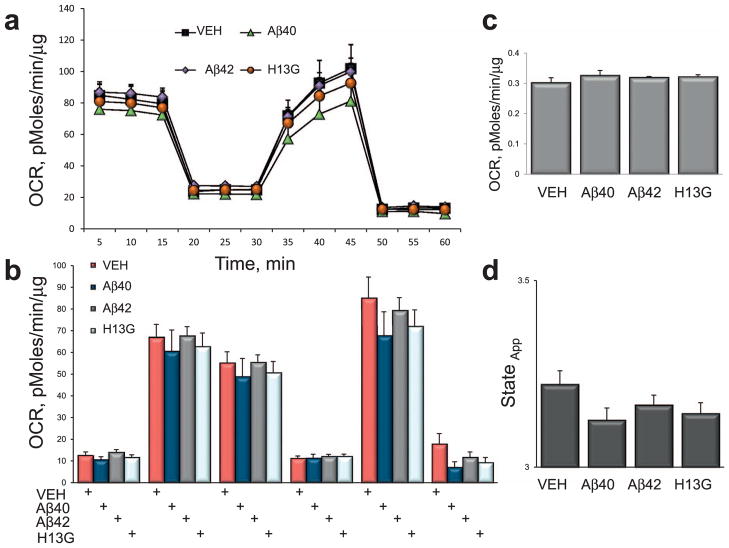
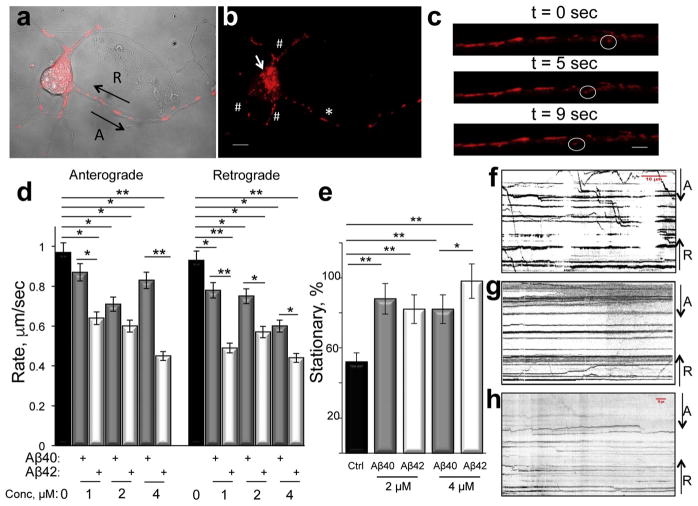
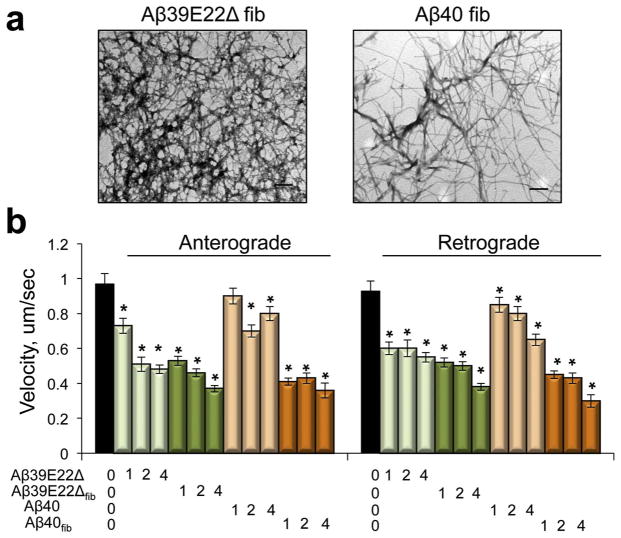
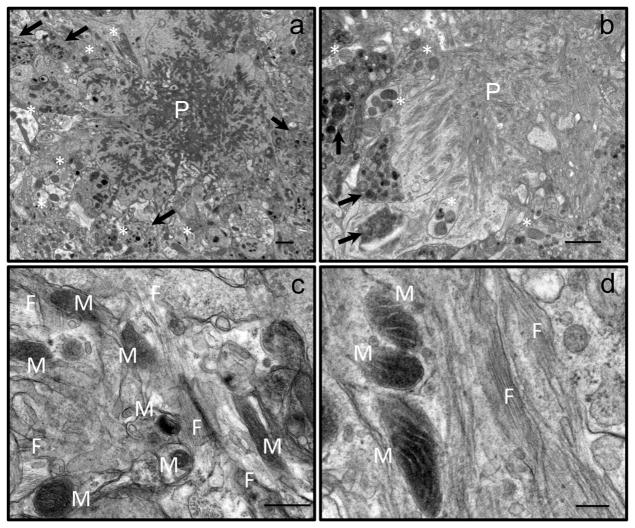
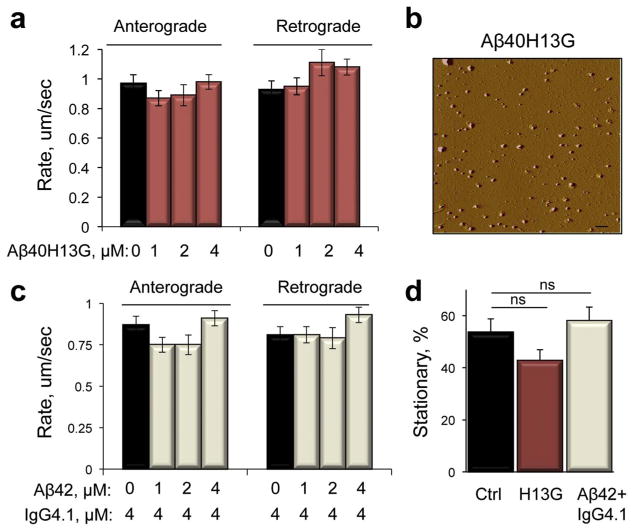
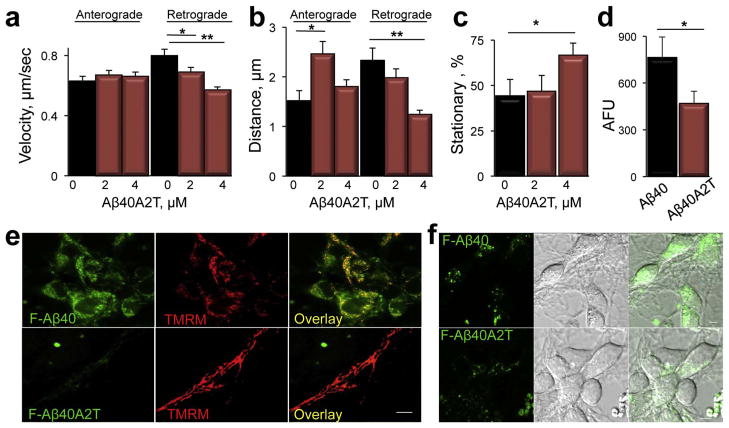
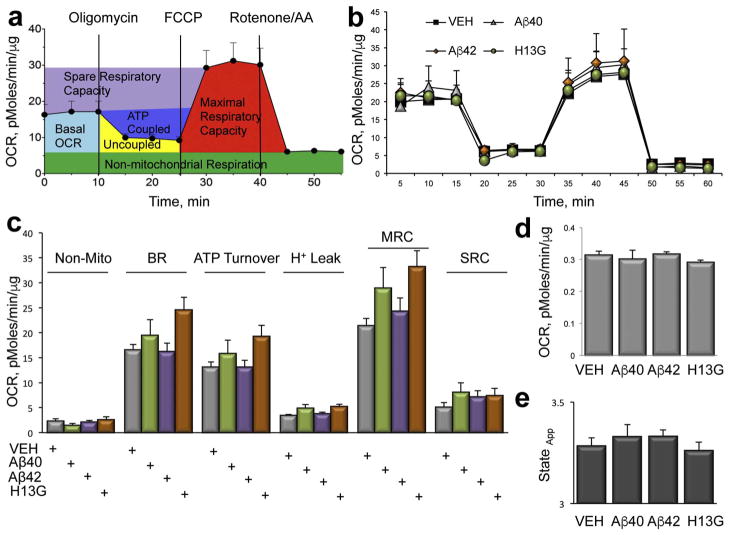
Inhibition of mitochondrial axonal trafficking by amyloid beta (Aβ) peptides has been implicated in early pathophysiology of Alzheimer's Disease (AD). Yet, it remains unclear whether the loss of motility inevitably induces the loss of mitochondrial function, and whether restoration of axonal trafficking represents a valid therapeutic target. Moreover, while some investigations identify Aβ oligomers as the culprit of trafficking inhibition, others propose that fibrils play the detrimental role. We have examined the effect of a panel of Aβ peptides with different mutations found in familial AD on mitochondrial motility in primary cortical mouse neurons. Peptides with higher propensity to aggregate inhibit mitochondrial trafficking to a greater extent with fibrils inducing the strongest inhibition. Binding of Aβ peptides to the plasma membrane was sufficient to induce trafficking inhibition where peptides with reduced plasma membrane binding and internalization had lesser effect on mitochondrial motility. We also found that Aβ peptide with Icelandic mutation A673T affects axonal trafficking of mitochondria but has very low rates of plasma membrane binding and internalization in neurons, which could explain its relatively low toxicity. Inhibition of mitochondrial dynamics caused by Aβ peptides or fibrils did not instantly affect mitochondrial bioenergetic and function. Our results support a mechanism where inhibition of axonal trafficking is initiated at the plasma membrane by soluble low molecular weight Aβ species and is exacerbated by fibrils. Since trafficking inhibition does not coincide with the loss of mitochondrial function, restoration of axonal transport could be beneficial at early stages of AD progression. However, strategies designed to block Aβ aggregation or fibril formation alone without ensuring the efficient clearance of soluble Aβ may not be sufficient to alleviate the trafficking phenotype.
淀粉样蛋白β (Aβ) 肽对线粒体轴突运输的抑制作用与阿尔茨海默病 (AD) 的早期病理生理学有关。然而,目前尚不清楚运动能力的丧失是否必然导致线粒体功能的丧失,以及轴突运输的恢复是否代表一个有效的治疗靶点。此外,虽然一些研究将 Aβ 寡聚物鉴定为运输抑制的罪魁祸首,但其他研究则提出原纤维发挥了有害作用。我们已经研究了在原代皮质小鼠神经元中具有不同突变的一系列 Aβ 肽对线粒体运动的影响。具有更高聚集倾向的肽会更大程度地抑制线粒体运输,而原纤维则会引起最强的抑制作用。Aβ 肽与质膜的结合足以诱导运输抑制,其中与质膜结合和内化减少的肽对线粒体运动的影响较小。我们还发现,具有冰岛突变 A673T 的 Aβ 肽会影响线粒体在轴突中的运输,但在神经元中其与质膜结合和内化的速率非常低,这可以解释其相对较低的毒性。Aβ 肽或原纤维引起的线粒体动力学抑制不会立即影响线粒体的生物能量和功能。我们的结果支持这样一种机制,即可溶性低分子量 Aβ 物种在质膜处起始轴突运输抑制,并被原纤维加剧。由于运输抑制与线粒体功能丧失并不一致,因此在 AD 进展的早期阶段恢复轴突运输可能是有益的。然而,单独设计用于阻止 Aβ 聚集或原纤维形成而不确保有效清除可溶性 Aβ 的策略可能不足以缓解运输表型。